
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2025-05-25
浏览次数:0
1.2、安装
如果后期有融合基因等需求,一定要注意版本。
[]
可以下载源码自行编译安装。STAR仅依赖最基础的gcc库。
## 适用于Ubuntusudo apt-get updatesu[id_906044060] apt-get install g++sudo [id_1287817955]## 适用于Red Hat, CentOS和Fedorasudo yum updatesudo yum install makesudo yum install gcc-c++sudo yum install glibc-static## 适用于SUSEsudo zypper updatesudo zypper in gcc gcc-c++wget 请访问该链接以获取STAR软件的2.7.1a版本,该版本已存档于GitHub上:https://github.com/alexdobin/STAR/archive/2.7.1a.tar.gz。tar -xzf 2.7.1a.tar.gzcd STAR-2.7.1amake STAR
1.2.2、conda安装
conda install -c bioconda star ## 默认安装conda上的最新版1.3、基本流程
STAR的基本流程包括两步:
创建基因组索引:用户需提交基因组参照序列,即FASTA格式的文件dnastar序列比对,以及相应的注释文件,格式为GTF。这一过程只需进行一次,构建完成后,即可应用于后续的比对分析。
将reads比对到基因组上。
2、构建基因组索引2.1、基本参数
STAR 启动线程N,线程总数为NumberOfThreads。--runMode genomeGenerate \基因组目录位于路径/to/genomeDir位置。请指定基因组fasta文件,例如:/路径/至/基因组/fasta1,/路径/至/基因组/fasta2,以此类推。运行sjdbGTFfile命令,指定/path/to/annotations.gtf作为文件路径。--sjdbOverhang ReadLength-1
参数说明:
也可以下载构建好的索引
wget请访问以下链接获取所需资源:https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/,其中包含了GRCh38_gencode_v33_CTAT_lib_Apr062020版本的插件式库文件,该文件已打包于名为plug-n-play.tar.gz的压缩包中,更新日期为2020年4月6日。tar该文件名为“zxvf GRCh38_gencode_v33_CTAT_lib_Apr062020.plug-n-play.tar.gz”,系禁止修改的专用资源。
2.2、构建索引时应该包括哪些//
这里涉及到几个定义
:指在染色体组装好后的更新序列(相当于更新包)。包括两种:
指的是这些序列版本存在于基因组特定区域,与最初的基因组组装序列保持平行状态,通常在众多个体间被发现,它们被视为对原始参考基因组的一种补充材料。
在创建索引的过程中,宜涵盖核心染色体,例如人类中的1号至22号染色体、X染色体、Y染色体以及线粒体染色体,同时亦应包含那些尚未明确定位的染色体。虽然这些染色体在构建索引时对整体大小影响微乎其微,但在实际进行序列比对时,却会有大量的rRNA序列与之匹配。若在构建过程中遗漏了这些序列,则这些读段可能被视为未与基因组进行比对,甚至可能被错误地标注在基因组的其他区域。
但是,在构建索引时,最好不要包括和。
也就是说,在建立索引的过程中,建议优先选用那些带有PRI()字段的*.dna..文件。
3、fastq文件比对到基因组3.1、STAR命令参数
mkdir 5.mappingcd ./5.mapping/将位于用户家目录下的4.trimg文件夹中的所有.fq.gz格式的文件,通过符号链接的方式,链接到当前目录下。cat ../SRR_Acc_List.txt | while read iddoecho -n "STAR --runThreadN 12 "echo -n 请勿对以下路径进行修改:"~/reference/linux/STAR/STAR_GRCh38_genecode_v33/ref_genome.fa.star.idx/",该路径指向了STAR软件使用的参考基因组索引文件。echo -n 禁止输出类型为BAM的已排序坐标数据,同时禁止输出未映射的原始快速序列数据。echo -n 使用quantMode参数进行基因计数分析,指定readFilesCommand为zcat,并采用Basic模式的二遍处理。echo -n 使用“BySJout”进行输出过滤,并设定“--outFilterMultimapNmax 20”来限制最大多映射数量为20。echo -n 设置输出过滤条件为最大误匹配数不超过999,同时确保误匹配率超过读段长度的0.04时才进行过滤。echo -n 设置对齐首尾的最小重叠为8,同时将对齐首尾的动态边界最小重叠设为1。echo -n 设置 chimSegmentMin 为 20,chimJunctionOverhangMin 同样设为 20,chimOutJunctionFormat 则指定为 1。echo -n 设置内含子最小长度为20,内含子最大长度为1000000,配对体间最大间隔为1000000。echo -n 设置chimSegmentReadGapMax为0,同时调整alignSJstitchMismatchNmax的值为-1,其余参数均保持为0。echo "--readFilesIn ${id}_rm_1_val_1.fq.gz ${id}rm_2_val_2.fq.gz 文件输出时,请指定文件名前缀。${id}"done > star.shless star.shnohup bash star.sh &
3.2、参数分块解读3.2.1、比对定量部分
STAR --runThreadN 12 # 12线程--genomeDir ~/reference/linux/STAR/STAR,基于GRCh38参考基因组,版本为gene_code_v33,索引文件位于ref_genome.fa.star.idx目录下。# 参考基因组索引所在位置禁止对序列进行修改,确保输出为按坐标排序的BAM格式。# 输出经过坐标排序的BAM文件--outReadsUnmapped Fastx # 输出没能比对到基因组上的序列,格式与输入文件相同对基因计数和转录组SAM文件的处理,需遵循特定的模式,不得擅自更改。生成基因表达水平的Read Count文件和转录本丰度测量的SAM文件。--readFilesCommand zcat # 输入的fastq文件经过gzip压缩--twopassMode Basic # STAR特有,两次对比模式执行读取操作,针对文件 ${id}_1_val_1.fq.gz 和 ${id}_2_val_2.fq.gz,进行数据加载。# 输入文件的名称--outFileNamePrefix ${id} # 输出文件的前缀以下参数的设定源自ENCODE官方,其中部分内容在中文中难以找到准确的对应翻译,具体可参考下方的插图。--outFilterMultimapNmax 20 若一段阅读材料被多次进行比对且比对次数超过20次,那么该阅读材料便不再被视为可与基因组进行匹配。--outFilterMismatchNmax 999 # 每对读段允许错配999个碱基(相当于不过滤)禁止超出读取长度上限的过滤不匹配数0.04 每对阅读段中,允许存在不超过其长度的4%的碱基不匹配,以PE150为例,这意味着每150个碱基中,最多可以有2*150*0.04=12个碱基出现错配。--alignIntronMin 20 # 内含子最短是20个碱基--alignIntronMax 1000000 # 内含子最长是1000000个碱基--alignMatesGapMax 1000000 # 一对读段之间最长距离为1000000个碱基
3.2.2、可变剪切部分
## 以下参数设置来自ENCODE官方,有些解释很难翻译成中文,参见下图--outFilterType BySJout # 对junction进行过滤以减少错误--alignSJoverhangMin 8 未经注释的连接区域的最低overhang长度为8个碱基。--alignSJDBoverhangMin 1 对注释过的连接点而言,其最短的外延长度仅为1个碱基。## 其他参数将最大缝接偏差Nmax设置为0,将偏差修正参数设置为-1,同时保持其他参数为0。设定剪切点错配的最大数量,其中“-1”表示不受限制。这四个数字依次对应于:(1)非典型剪切;(2)GT/AG或CT/AC;(3)GC/AG或CT/GC;(4)AT/AC或GT/AT。
3.2.3、融合基因部分
--chimSegmentMin 20 每对嵌合的阅读片段中较短的那一端需包含至少20个碱基,换言之,PE150规则下,融合基因的结构至少为280加上20个碱基。--chimOutJunctionFormat 1 输出的Chimeric.out.junction文件可直接使用,无需进一步处理,即可用于分析融合基因。--chimSegmentReadGapMax 0 # 嵌合读段之间不允许空位--chimJunctionOverhangMin 20 嵌合的接头区域的最小悬突长度为20个碱基,这一设置旨在筛选掉那些过于短暂的外显子,从而排除连续剪切事件。
4、STAR参数图解
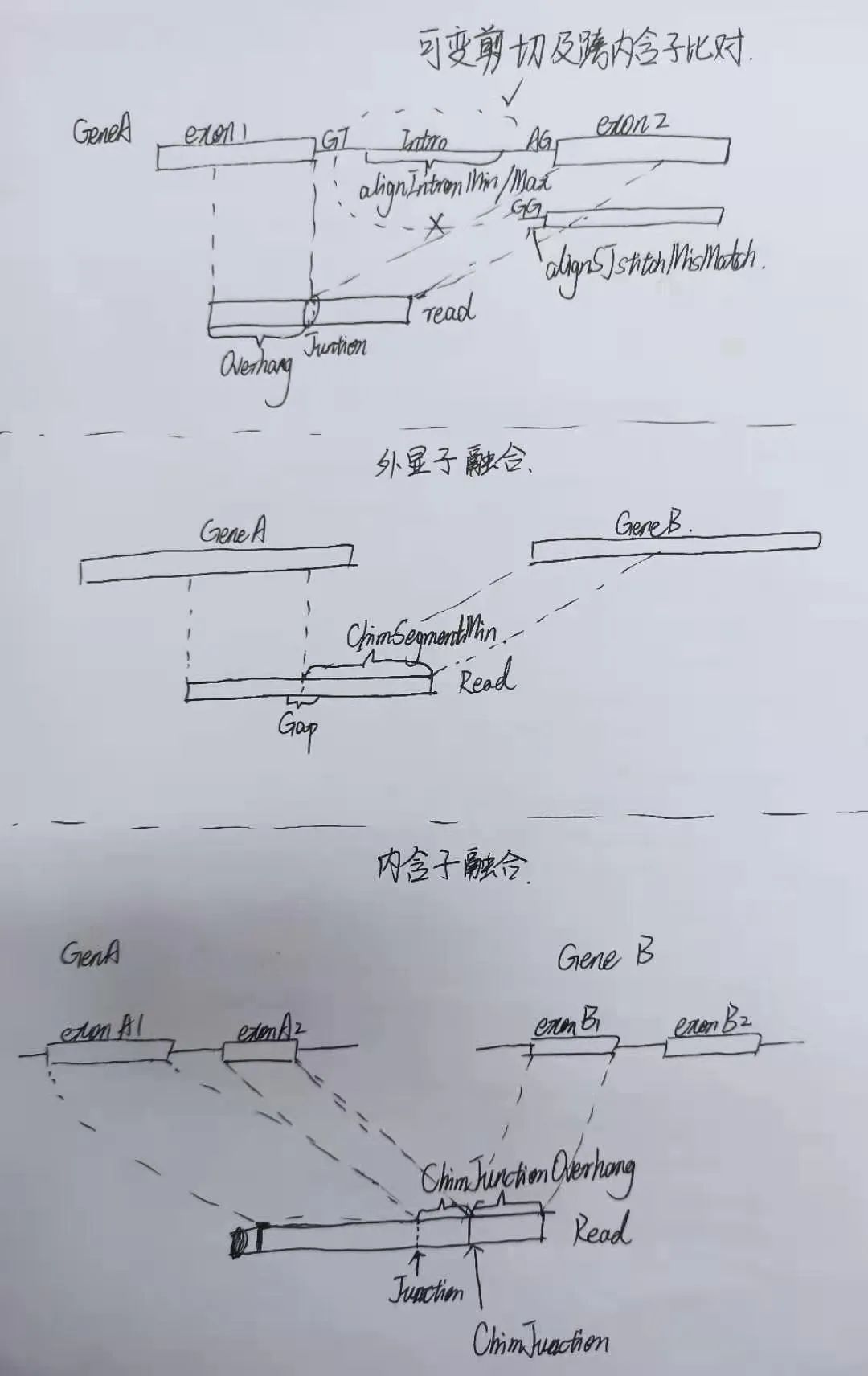
凑合着看吧。。。
目前,比对定量工作已圆满结束dnastar序列比对,在下一期,我们将着手对STAR生成的输出文件进行详细解读。
文末友情推荐
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码