
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-06-06
浏览次数:0
完成以上操作后,就可以进行量化了。 对于参考转录组,通常采用比较和量化的方法。 在这里,我使用 STAR 进行量化。
一、STAR介绍与安装 1.1. 介绍
STAR全称toadnastar序列比对,是著名项目使用的RNA-seq比对软件。 STAR使用底层C++语言编译,可在多核上运行,比对速度极快。 与其他两款常用的参考转录组比对软件相比dnastar序列比对,具有更高的独特比对率。 与GATK的良好兼容性使得RNA-seq更容易发现基因突变。 据悉,10X的单细胞转录组上游软件也是基于STAR的。
下载地址为; 创建的索引文件和可以用来建立索引的文件的下载地址是。 值得注意的是,本站索引仅适用于star2.7.4a,其他版本需要自行创建。
1.2. 安装
如果后期有融合基因等需求,一定要注意版本。
1.2.1、自行编译
可以下载源码自行编译安装。 STAR 只依赖于最基本的 gcc 库。
## 适用于Ubuntusudo apt-get updatesudo apt-get install g++sudo apt-get install make## 适用于Red Hat, CentOS和Fedorasudo yum updatesudo yum install makesudo yum install gcc-c++sudo yum install glibc-static## 适用于SUSEsudo zypper updatesudo zypper in gcc gcc-c++wget https://github.com/alexdobin/STAR/archive/2.7.1a.tar.gztar -xzf 2.7.1a.tar.gzcd STAR-2.7.1amake STAR
1.2.2、conda安装
conda install -c bioconda star ## 默认安装conda上的最新版1.3. 基本流程
STAR的基本过程包括两个步骤:
基因组索引创建:在这一步中,用户需要提供基因组参考序列(FASTA文件)和注释文件(GTF文件)。 它只需要创建一次,就可以用于所有后续的比较。
将读数与基因组对齐。
2.基因组索引的构建 2.1. 基本参数
STAR --runThreadN NumberOfThreads \\--runMode genomeGenerate \\--genomeDir /path/to/genomeDir \\--genomeFastaFiles /path/to/genome/fasta1 /path/to/genome/fasta2 ... \\--sjdbGTFfile /path/to/annotations.gtf \\--sjdbOverhang ReadLength-1
参数说明:
您也可以下载创建的索引
wget https://data.broadinstitute.org/Trinity/CTAT_RESOURCE_LIB/GRCh38_gencode_v33_CTAT_lib_Apr062020.plug-n-play.tar.gztar -zxvf GRCh38_gencode_v33_CTAT_lib_Apr062020.plug-n-play.tar.gz
2.2. 建立索引时应该包括什么//
这里有几个定义
:指染色体组装后的更新序列(相当于更新包)。 包括两种类型:
: 指基因组某一区域不同版本的序列,与原始基因组组装序列平行,常存在于不同个体中,可以看作是对原始参考基因组的补充
索引时最好包括主要染色体(人类的 chr1-22、chrX、chrY 和 chrM)和未映射的染色体。 建索引的时候加入这个pair与索引的大小无关,实际比对的时候会有大量的rRNA序列比对。 如果这些序列未包含在构建中,则此类读取将被视为未映射到基因组,甚至错误映射到基因组中的其他位置。
但是,在建立索引时,最好不要包括和。
也就是说,在 *.dna.. 中标有 PRI() 数组的文件被推荐用于索引。
3. Fastq 文件与基因组的比较 3.1。 STAR 命令参数
mkdir 5.mappingcd ./5.mapping/ln -s ~/path/to/4.trimg/*.fq.gz ./cat ../SRR_Acc_List.txt | while read iddoecho -n "STAR --runThreadN 12 "echo -n "--genomeDir ~/reference/linux/STAR/STAR_GRCh38_genecode_v33/ref_genome.fa.star.idx/ "echo -n "--outSAMtype BAM SortedByCoordinate --outReadsUnmapped Fastx "echo -n "--quantMode GeneCounts --readFilesCommand zcat --twopassMode Basic "echo -n "--outFilterType BySJout --outFilterMultimapNmax 20 "echo -n "--outFilterMismatchNmax 999 --outFilterMismatchNoverReadLmax 0.04 "echo -n "--alignSJoverhangMin 8 --alignSJDBoverhangMin 1 "echo -n "--chimSegmentMin 20 --chimJunctionOverhangMin 20 --chimOutJunctionFormat 1 "echo -n "--alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 1000000 "echo -n "--chimSegmentReadGapMax 0 --alignSJstitchMismatchNmax 0 -1 0 0 "echo "--readFilesIn ${id}_rm_1_val_1.fq.gz ${id}_rm_2_val_2.fq.gz --outFileNamePrefix ${id}"done > star.shless star.shnohup bash star.sh &
3.2. 参数块分析 3.2.1. 定量比较
STAR --runThreadN 12 # 12线程--genomeDir ~/reference/linux/STAR/STAR_GRCh38_genecode_v33/ref_genome.fa.star.idx/ # 参考基因组索引所在位置--outSAMtype BAM SortedByCoordinate # 输出经过坐标排序的BAM文件--outReadsUnmapped Fastx # 输出没能比对到基因组上的序列,格式与输入文件相同--quantMode GeneCounts TranscriptomeSAM # 输出基因的Read Count文件以及转录本定量的SAM文件--readFilesCommand zcat # 输入的fastq文件经过gzip压缩--twopassMode Basic # STAR特有,两次对比模式--readFilesIn ${id}_1_val_1.fq.gz ${id}_2_val_2.fq.gz # 输入文件的名称--outFileNamePrefix ${id} # 输出文件的前缀## 以下参数设置来自ENCODE官方,有些解释很难翻译成中文,参见下图--outFilterMultimapNmax 20 # 如果一个读段被多重比对超过20次,则认为这个读段不能被比对到基因组--outFilterMismatchNmax 999 # 每对读段允许错配999个碱基(相当于不过滤)--outFilterMismatchNoverReadLmax 0.04 # 每对读段允许出现读长*4%的碱基错配,即PE150允许2*150*0.04=12个碱基错配--alignIntronMin 20 # 内含子最短是20个碱基--alignIntronMax 1000000 # 内含子最长是1000000个碱基--alignMatesGapMax 1000000 # 一对读段之间最长距离为1000000个碱基
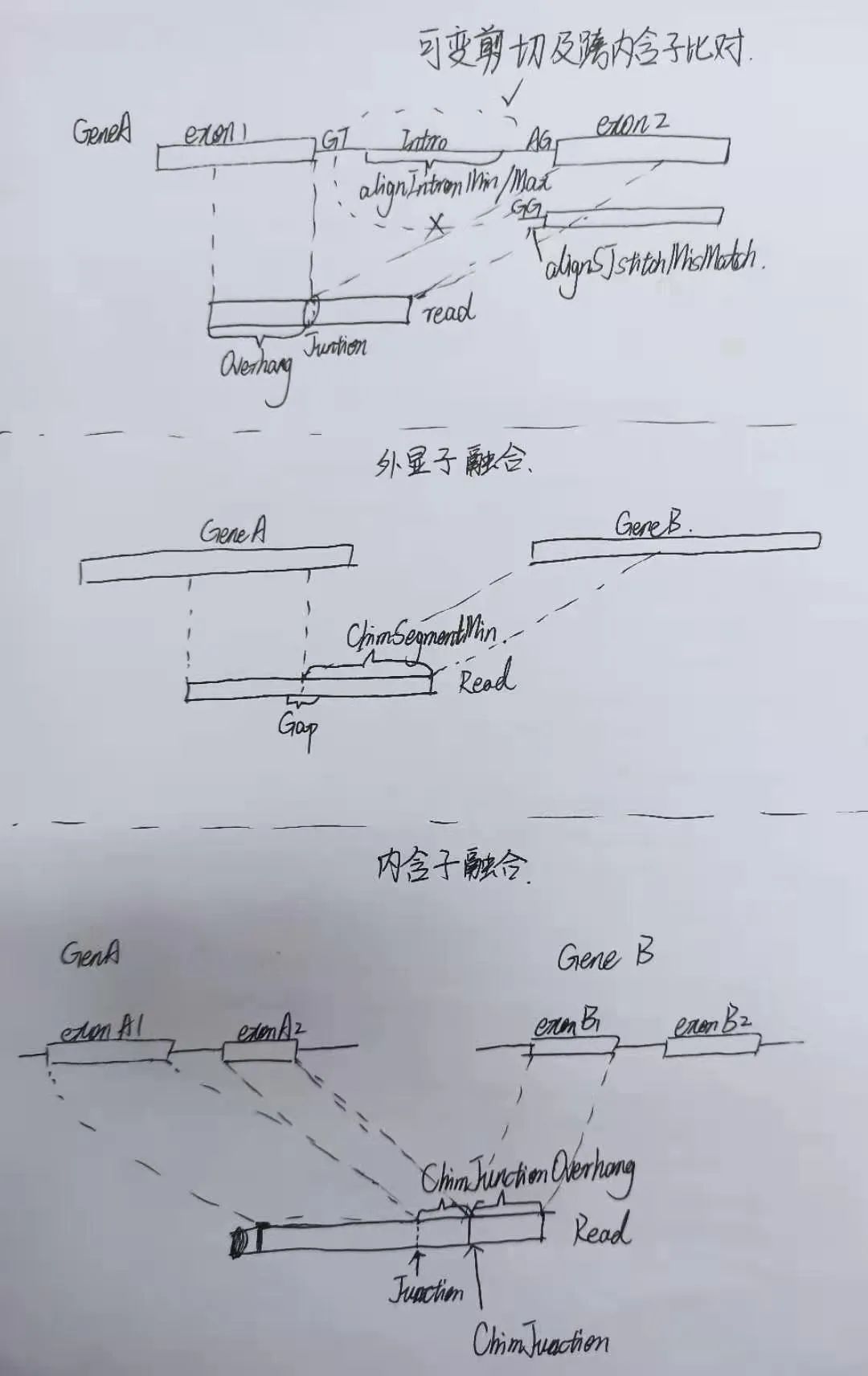
3.2.2、可变剪切部分
## 以下参数设置来自ENCODE官方,有些解释很难翻译成中文,参见下图--outFilterType BySJout # 对junction进行过滤以减少错误--alignSJoverhangMin 8 # 未注释过的junction的最少的overhang是8个碱基--alignSJDBoverhangMin 1 # 注释过的junction的最少的overhang是1个碱基## 其他参数--alignSJstitchMismatchNmax 0 -1 0 0 # 允许剪切点错配的个数(-1代表无限制)四个数字分别代表(1)非经典;(2)GT/AG或CT/AC;(3)GC/AG或CT/GC(4)AT/AC或GT/AT
3.2.3. 融合基因部分
--chimSegmentMin 20 # 每对嵌合读段较短的一端至少有20个碱基,即PE150允许280+20结构的融合基因--chimOutJunctionFormat 1 # 输出的Chimeric.out.junction文件可直接用于融合基因--chimSegmentReadGapMax 0 # 嵌合读段之间不允许空位--chimJunctionOverhangMin 20 # 嵌合的junction的最少的overhang是20个碱基,为了过滤非常短的外显子,即连续剪切事件
4.STAR参数图
等着瞧吧。 . .
现在对比量化已经完成,下期我们将对STAR的输出文件进行回顾分析
文末友情推荐
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码