
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-07-10
浏览次数:0
好吧,我还是成功地用标题吸引了你。
虽然,这个称号只是一个旗帜。
这是我第一次写这些非教程推文(可能不会是最后一次)。
明天我们给大家介绍一款非常小的(不知道有多小)的生活信软件。
嗯,这似乎是另一个软件教程。 。 。
毕业季,我们总是匆匆告别。 许多盛信软件的忠实(特殊)用户(粉丝)也毕业了。 与生活必需品QQ、微信相比,如果毕业生不再从事相关领域的研究,就意味着这种软件将失去这些曾经一起努力的同学,即使他们以前有过。
就失去了这么一位资深用户——课题组里做湿实验的Y师弟。 每次Y师弟对我的产品提出新的要求时,我总是表现出不屑(不屑)(困难)和专注(实现)。 而每次Y师弟说“师弟,你要珍惜我这个唯一的用户”,总是让我乖乖跑去折腾代码。
一开始我使用的是内置的接口包,可以满足大部分功能,但是有一点很不方便。 每次都要点击按钮导出输入文件(好像大部分软件都是这样)。 而对于科研任务极其繁忙的Y师弟(笔记本里的文件很多)来说,每次在哪个弹窗中查找文件都需要很长时间。 于是Y师弟就问是否支持拖拽文件导出?
不只是Y师弟,我也觉得它太弱了,没有拖拽文件的功能。 而且查了很久资料,发现这个框架其实很简单易学,适合入门。 不过,以后要实现文件拖拽虽然不容易,虽然能实现,但是代码也不好写。 另外,我还希望有一个界面提示文字,即键盘连接到某个按钮或者输入框等控件,它可以显示我想要告诉用户的内容。
所以,我决定改变框架(使用PyQt5)并重新打包。
不过,虽然我没有时间重画,但是我已经直接通过脚本实现了很多紧急任务,并且将脚本封装到界面中需要很长时间。 除了把接口写进代码之外,还需要写出用户容错风暴,需要不断地调试。
直到疫情期间,我终于有时间做这件事了。 于是,我学习了PyQt5的使用,并应用到了它上面。 我从头开始重新绘制它并添加了新功能。 最大的改变是在部分功能的实现上增加了多线程机制。
可惜Y师弟焕然一新的时候就已经毕业离开了。 。 。
说了这么多,好像还没介绍呢?
它是一款生物信息学的小软件,用于基因序列数据的处理,特别是病毒基因组的变异分析。 1.0版本开发已于2020年4月完成,目前仍在更新和建立中(非常欢迎需求或建议)。
现在,我们来翻译一下该软件目前可用的说明书,看看它有哪些功能。
说实话,这是我第一次先把英文手册翻译成中文,然后再翻译回来。 。 。
1 软件下载地址
该软件可免费下载并免费使用:
引用参考文献(目前仅预印本):
2 弱函数 2.1 序列格式转换
其实很多生物信息软件都有这个共同的功能,所以它也有。
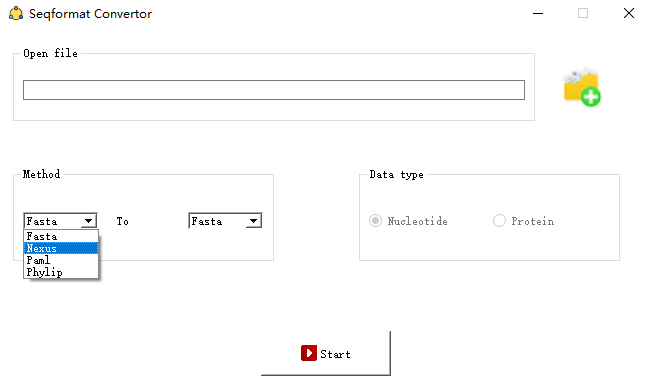
通过拖放(推荐)或按键直接导出基因序列文件,可进行不同格式之间的相互转换。 目前可相互转换的格式有Fasta、Nexus、Paml。 仅当目标格式为“Nexus”时,“序列类型(数据类型)”选项才可用。
那么将Fasta格式转换为Fasta格式有什么用呢? 最大的好处就是将非标准的fast格式(比如占用多行的序列)转换为标准的fasta格式(对于每个序列,序列名称占用一行,而序列只占用一行)。
2.2 序列变体
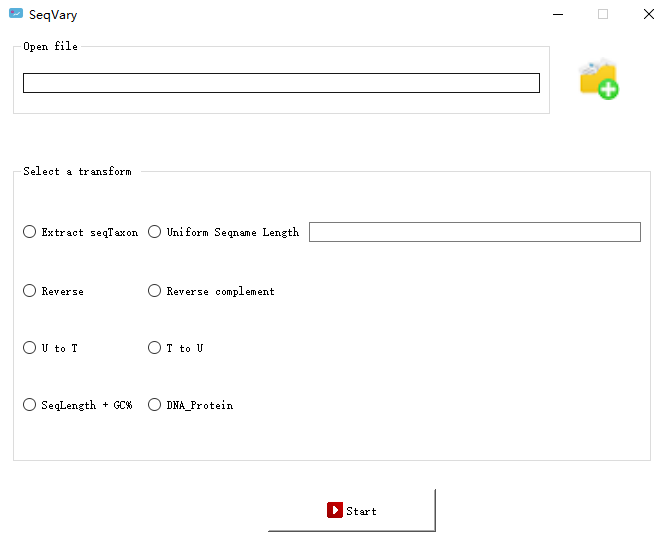
该接口共有8个子功能,分别是提取序列名()、统一序列名宽度()、序列反向()、序列反向补全()、RNA转DNA序列(UtoT)、DNA转RNA序列(TtoU) ),统计序列宽度和GC浓度(+GC%),DNA和蛋白质对照表()。
注意,如果选择“”选项处理基因序列,则要求输入序列是编码区的基因,并根据密码子形式进行比对。 至于这个功能是做什么的,大家自己尝试一下就知道了。
2.3 序列名称重命名
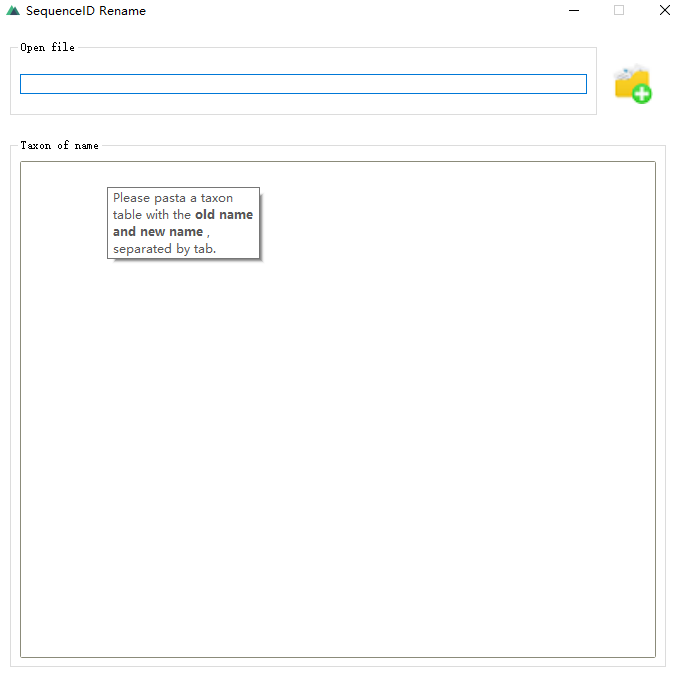
功能就像标题一样,不过这个名字虽然可以有多个断句,但是适用范围也不同(序列“名字重命名”、“序列名字”重命名)。 我更喜欢用它来处理树文件而不是处理序列。 特别是对于用于发表的进化树图片,我们一般需要按照一定的格式对树的分类单元进行统一命名。 因此,首先直接批量替换树文件中的序列名称,省去后面使用矢量图形工具(例如AI)的麻烦。 做出改变的麻烦。
2.4 剪切序列片段
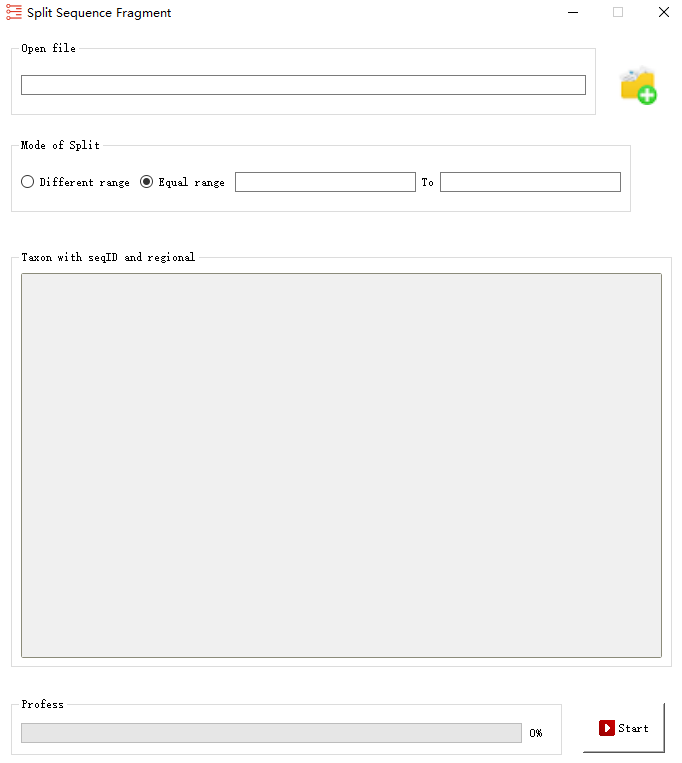
该函数用于批量截取指定范围内的基因片段,有两种不同的模式:每个序列指定一个范围(range)截取,以及等距截取()。 如果选择“范围”,则需要在文本框中粘贴每个序列对应的信息:序列名称、起始位置和结束位置,以制表符分隔,每个序列的信息抢占文本框中的一行。 如果是等距拦截,如上图。
2.5 串联基因

该函数用于将多个基因序列连接成一个。 您可以先将不同基因的序列数据(fasta格式)倒入同一个文件夹中,然后将文件夹推入输入框,然后点击“”,软件会将每个基因对应的序列文件路径导出到文本框中。 可以通过自动调整文本框中文件路径的顺序来更改基因的顺序。
2.6 基因的可视化提取
使用此功能之前,必须先将其安装在笔记本电脑上,然后点击菜单栏上的“基因”按钮,会出现如下界面:
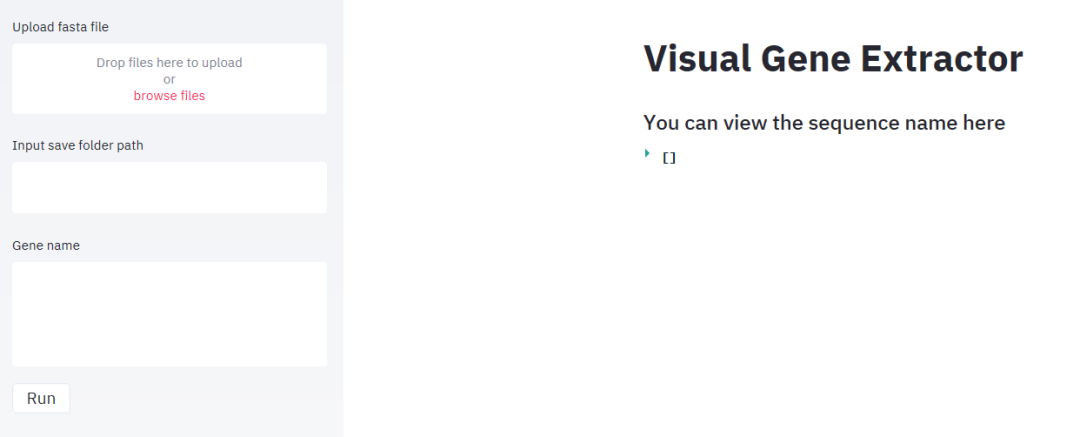
这个功能以本地化网页工具的形式呈现,因为我觉得网页模式很酷。 怎么用,我举一个反例。 我突然想起来这条推文是我以前写过的,所以看看:
没错,当时我把这个推文前面的脚本给了,还有很多做湿实验的朋友不会用,所以我就建了这个函数,做成了一个界面,现在直接就可以搞定了通过单击鼠标。 这个功能的本质就是通过提供的基因名称列表,从序列名称中批量匹配提取对应的序列,具体看它如何灵活操作,相信用户比我更会用。
3 勉强能用
下面第二部分的功能纯粹是用来陪衬的,接下来我会介绍一些勉强有道理的功能。
3.1 快速注释
为什么要写这个函数呢?
NCBI上的很多基因组序列都没有注释,我们在科学研究中有时需要用到这样的未注释序列,尤其是上面的某个基因序列。 言外之意就是我们必须对自己上传的序列进行注释。 是的,这些序列不是你的。 (强烈建议研究人员在将每个序列提交给 NCBI 时对其进行注释)。
序列注释的软件有很多,就不一一列举了。 无论是本地化软件还是在线软件,基本原理都是相似的。 首先,找到带有注释信息的参考序列。 这个序列需要和你要注释的序列相同 序列整体比较相似,然后放在一起进行多序列比对,根据参考序列的注释信息进行比对,然后确定(截取)的基因区域你的顺序。
标注单个序列还可以dnastar序列比对,任何软件都可以任意操作,估计量也不大。
当需要注释多个甚至数千、数万个序列时,问题当然会复杂一些。
在我这几年的研究生涯中,当遇到需要注释几十个甚至更多同源序列或者从大量未注释的全基因组序列中提取一个基因序列时,我一般采用以下实验方法:先下载一个或更多.gb格式的参考序列(带注释信息),与不带注释的序列(自己实验室下载或扩增的)一起拖入本软件中,然后使用内置的序列比对工具(如如Mucle或MAFFT),然后根据参考序列的注释信息,可以对这些未注释的全基因组序列进行注释或者截取所需的基因片段。

(参考序列与待注释序列比对后的部分截图)
当需要注释的序列很少时,这很方便。
但当序列较多时,也会出现问题。 当我处理从它下载的3000多个未注释的新冠序列时,执行序列比较实际上非常慢,即使它在服务器上运行。 如上所述,它是内置的或者是序列比较工具,例如Mucle。 说白了,Mucle本身就是一个独立的可执行程序(干过这个的人都应该知道),用这个独立的程序直接比较速度更快。
然而,如果使用单独的程序进行比对,虽然无法与参考序列的注释信息关联起来。 虽然我知道也有一个在命令行运行的注释程序,但这里就不介绍了。
于是,写了一个简单的功能,还是实用的。
原理很简单。 由于序列比对工具非常多,而且功能也比较完善,我们可以先用它们来进行多序列比对,所以关键点就是将待注释的序列与参考序列关联起来。 而且,如上所述,注释必须以同源性高的序列为参考。 所以,你可以这样做:
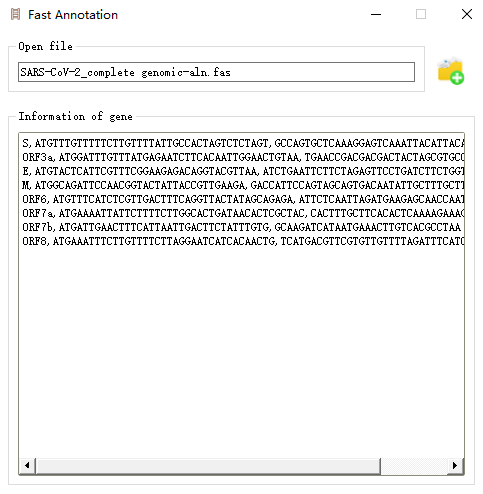
对于来自相同或高度相关物种的那些菌株(或弧菌)序列,核酸同一性通常相对较高。 为此,序列注释可以基于与参考序列基因信息的多重序列比对。 提供快速序列注释功能,通过导出已与其他软件多序列比对的全基因组序列集(fasta格式),并将用于注释的参考序列调整到文件顶部。 将参考序列对应的基因名称、起始字符串和结束字符串粘贴到文本框中,并用“,”分隔,即可批量注释基因。 请注意,基因的起始字符串或结束字符串的宽度没有要求,并且该字符串必须且仅需要在参考序列中是唯一的。 据悉,序列之间的相似度越高,注释的准确性就越高。
如上所述,如何知道参考序列中哪些字符串是唯一的? 这个比较简单,用记事本或者++之类的文本工具,打开比对的全基因组序列,Ctrl+F,将字符串粘贴进去,然后搜索。
3.2 序列单位矩阵
在很多文章中,我们都可以看到核酸(nt)或者多肽(aa)的()矩阵(对于序列同一性和相似性()的联系和区别,请自行百度),比如下面这样:

(LiJin-Yan 等人,2020)
或者它显示核酸和肽的同一性,如下所示:
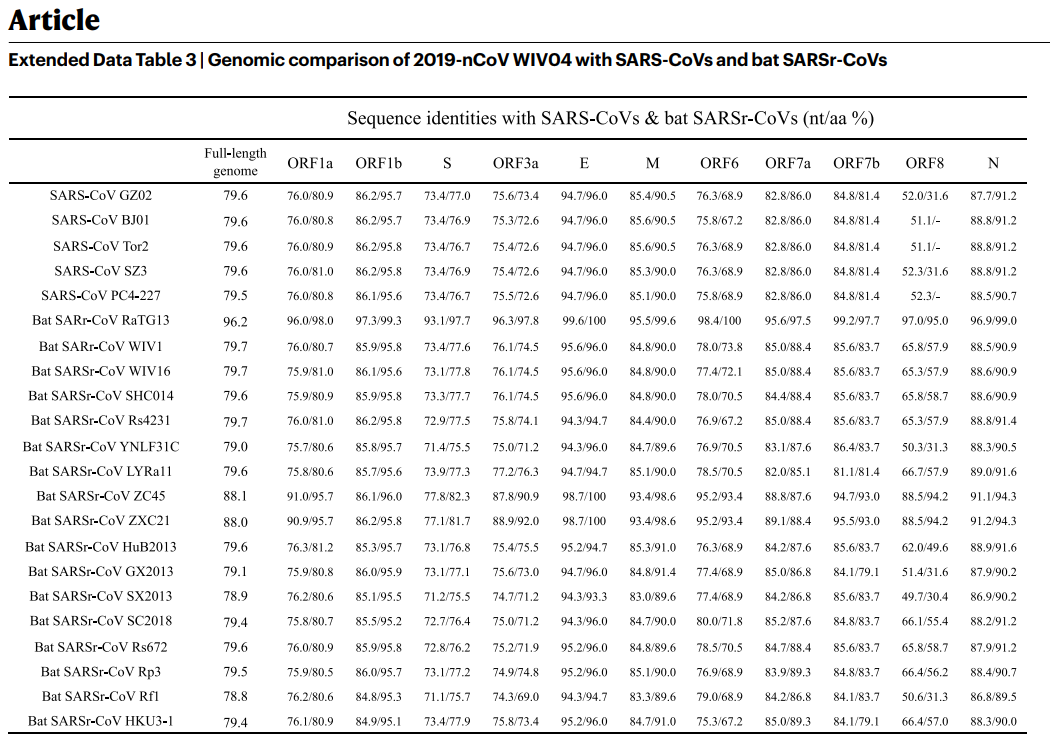
(周鹏等人,2020)
在它出现之前,我曾经使用这个软件来估计序列同一性。 但它有一个很大的局限性,就是之前的核酸+肽单位矩阵表,无法一步生成。 它首先必须单独估计核酸或肽身份表,然后自动(对于不懂编程的人)或通过脚本(对于懂编程的人)合并。
工欲善其事,必先利其器。
今天,这是完全可能的。
点击 中的菜单,出现下界面,然后推入按照标准密码子形式()比对的编码基因序列文件(也可以通过按键加载),选择“”。

然后,点击“开始”,直接输出表格(输出结果在输入文件对应的文件夹下):
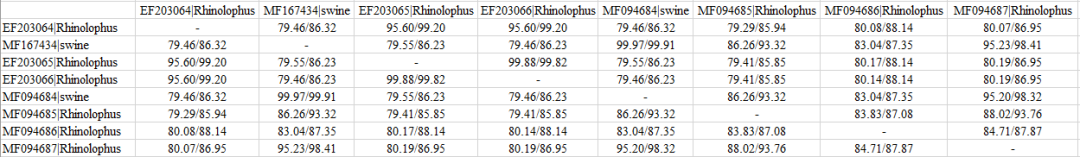
分析结果快速准确,表格稍加整理即可直接用于文章发表。 除此之外,它还提供了压缩间隙函数(gap),用于估计编码区基因核酸序列的同一性。 事实上,这是一个选择。 如果选中该选项,则比对序列中每3个连续插入或删除(缺口)的核苷酸将被视为1个,然后将估计成对序列同一性。 有生物学背景的朋友可以稍微思考一下。 通过这样做的原因。
3.3 去除高度相似的序列
虽然这条推文的标题是一个横幅(因为我懒得想标题),而且这个函数实际上存在于 .
在我们的科研工作中,有时会遇到两个需求:
(1) 消除相同的序列,只保留一个。 除此之外,有时您想知道哪些序列是相同的。 这里的相同是指两个序列的同一性或相似性为100%。
(2)虽然有些序列相似度低于100%,但它们可能是99%或更高,例如99.99%。 有时,当我们使用这个序列作为进化树中的参考序列时,我们可能只需要一个序列,而高度相似的则需要被淘汰。
那么,相似度达到多少才算是高度相似呢? 其实没有标准,也不需要标准,要看研究的背景和目的。 此外,需要消除高度相似的序列。
虽然网上有工具可以处理,而且我用过,觉得不是很好用。
因此,您可以再次执行此操作 (>-H-):
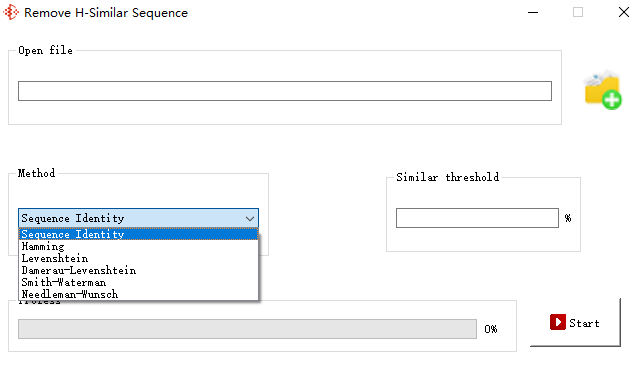
应该强调的是,提供了 6 种不同的算法来估计序列相似性。 需要注意的是,“”和“”距离方法要求输入序列已经被多个序列比对。 同时,也建议使用这两种方法来消除高度相似的序列。 因为速度比较快,所以特别推荐“”算法。 这个方法经过我大量的打磨。 对于剩下的4种方法,最好不要提前对输入序列进行比对,因为该算法具有序列比对功能。 用户可以通过指定阈值()来定义要消除多高相似度的序列。 请注意,输入阈值也包含在该范围内。
为此,如果将阈值设置为100,程序就会启动消除重复序列的功能。 此时,虽然无论选择哪种算法都是一样的,因为程序采用了另一种高效的处理机制。
这就是结局? 还没有,如果你在程序页面的空白键盘上点击右键,那么恭喜你,你已经启用了隐藏功能。 您可以使用这个隐藏函数来获得每个算法估计的序列相似度矩阵。 事实上,对于“”算法,确切地说是恒等式。 不再有截图进行演示。
4个不太好的功能
说实话,我总觉得有类似的软件可以实现上述的功能,但是我没有见过,也没有用过,很有可能是我在重新发明轮子(重新发明轮子是必然的,一波分析(如果需要用户在不同的软件之间来回切换就不好了),或者它们的功能不是比较健全。 其实有创新,但还不够,编程难度也不够。 不过,下面要提到的三个组合功能都经过了残酷的实践检验。
如果你能从头到尾听到这句话,那么你一定是铁粉,或者你很佩服推文的作者(偷偷观察……)。
让我们从复合函数中的第一个函数开始:
4.1 计分器
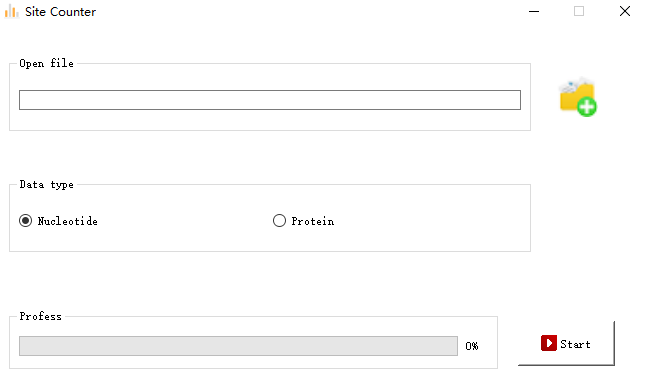
是的,又是这些极简风格。 虽然有时我想让界面变得更复杂,多一些按钮和参数设置框,但总觉得太复杂了,不利于用户快速上手。 根据我自己的经验,在分析和处理数据时,大部分时间都花在学习如何使用分析软件和处理程序错误上。 其实这也和我写的功能都是一些技术集中度太高的小功能有关。
言归正传,上图中的“Site”是做什么用的?
翻译过来就是“site ”,就是一个简单的统计功能。 正如软件手册中所写:
这可以是每个碱基(或氨基酸)的类型、数量和数量。
网站为. 在,威拉
基于每个位点的碱基(或氨基酸)。
分析结果以表格形式输出。 我比较后导出了碱基序列,运行程序进行分析后,我取出其中一份结果(*.csv),简单地用Excel自带的图画出来。 它看起来像这样:
横坐标是基因位点,纵坐标是浓度,颜色代表不同的核苷酸。
虽然我在一些高水平的SCI论文中看到过这些图片,但其实我并没有看到这种展示能带来的学术效果,而且足够漂亮和炫酷。
4.2 突变分析
生物体,尤其是病毒,尤其是RNA病毒,遗传变异是司空见惯的事情。 为此,我们经常使用比较基因组学。 再深入一点,基因组或单基因多序列比对的工具也很常见,例如MAFFT。
然而,有一件非常奇怪的事情。 虽然序列比对的工具很常见,但直接用于基因变异分析的下游软件却很少。 可能是因为这个功能往往比较简单,懂编程的大鳄用脚本就可以搞定,不需要做接口程序,而不懂编程的人,比如大多数湿实验者,是无法开发的它。
或者MEGA、、、(付费软件)等软件自带序列显示功能,也可以浏览变异位点的情况。 而且,少量的序列还可以,如果有几万个,虽然检查这些变异很不方便,尤其是在很长一段时间后。
疫情期间,我接到一个任务,要分析3000多条新冠序列(粗细左右)的突变状态,需要全面细致的分析,精确到每一个密码子、核苷酸位置,不仅是全基因组层面,而是也在全基因组水平上。 每个编码基因也需要进行分析(上面提到的快速注释功能就是在这样的条件下开发出来的),特别是要快速定位突变热点或热点,能够识别不同类型的突变(如同义突变、非同义突变)突变、提前终止),除此之外,还需要鉴定多肽的性质是否发生变化,最后分析结果需要高度概括,更重要的是要快! 。
虽然,我早已忘记了当时导师所说的“快”是催促我快速分析(自动或使用现有软件),还是暗示我要开发软件进行快速分析。 然而,经过深思熟虑,我搜索并没有找到合适的现有软件可以完全完成这项工作。 真的会自动处理吗?
果然,无论导师的意思是什么,我都只能选择前者。
“要做研究,就用最好的工具,如果没有,那就构建它”——这是我写软件的初衷和动机。
因此,使用以下函数:
该函数用于剖析单个编码基因或编码基因组合的突变特征。 输入的序列需要提前使用其他软件并基于标准密码子方法(需要fasta格式)进行比对,然后扫描序列数据集中的每个密码子位点,将序列集中的第一个序列用作参考序列。 识别 5 种不同的突变类型,包括同义、非同义、插入、删除和终止密码子,还可以标记简并核苷酸。
最后,所有的突变位点、突变类型以及对应位点的突变频率都会以表格的形式高度概括。 对于非同义替换位点,可以鉴定突变多肽的性质(例如极性或电荷)是否发生变化,这可以帮助我们识别一些重要的变异位点。 除此之外,还提供了一个单独的文件dnastar序列比对,标记了突变位点对应的突变菌株(弧菌),方便参考。
其实如果只有前面的功能的话,还是有点弱。
虽然我们知道有多少个突变位点、有哪些位点,但这还不够,因为有些位点的突变可能只出现在少数序列中(即突变频率较低),甚至有些位点的突变只出现在少数序列中(即突变频率较低)。出现在单个序列中(突变频率为1),可能是测序错误造成的。 为此,我们需要一些东西来帮助我们确定是否存在大规模的突变。
这个东西,最好有图片,因为这样更直观。
因此,提供一键突变频率分布图(仅包括同义突变位点和非同义突变位点)。 参数示例如上图所示,结果为矢量地图供发布。 文本框中的每一行代表一组替换频率,最小值和最大值由制表符分隔。 请注意,每组的最小值不包括在频率范围内。
需要注意的是,替换频率分布图是由R语言绘制的,因此要使用该功能,需要先在笔记本电脑上安装R,然后通过插件功能向其添加.exe程序。 如果.exe没有成功绑定,运行时会手动提示。
上述结果的一个例子如下:

(突变位点总结)

(突变位点+突变株)
(突变频率分布图)
4.3 站点过滤
关于这个函数,通过一个反例可以更好的理解。
例如,对上述突变进行分析后发现,第1314位点是突变热点,富含A和C两种核苷酸,分别占40%和60%。 现在需要提取该位点的所有核苷酸C序列以进行后续分析。
或者,你发现有3个位点突变频率比较高,而且突变的核苷酸总是互相出现,这有点像连锁不平衡遗传(具体百度一下),所以你根据这三个位点提取对应的序列。
所以,你可以这样做:

只需要以fasta格式导出序列集,在文本框中输入要提取的位点和核苷酸(或多肽),用英文冒号分隔,一行写出一个位点的信息,不同的站点只需将其写在不同的行上即可。
最后5句话
目前它还有限且简单,并将不断发展。 如果您有兴趣,请告诉我。 特别欢迎您提出需求或建议,需求带动生产。
我建了一个QQ群,供用户交流。 如果您需要反馈程序bug,或者询问需求,或者建议,或者其他等等,都可以加入。 组号:.
6 最后 最后
前面的故事似乎还没有讲完。 Y师弟离开中学前,第一件事就是用U盘从我的笔记本上拷贝最新版本,然后再拷贝其他的东西。 他很感动。
感谢所有直接或间接做出贡献或提出建议的人,即使你们没有直接参与开发,我为你们编写的大部分脚本都在其中使用。 目前还很小,未来还有很长的路要走。 它的功能扩展和建立需要您的贡献和建议。

如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码