
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-06-07
浏览次数:0
指导
考虑到比较转录组时,可用的软件很多,但很少介绍它们之间的差距。 为此,本文主要介绍STAR,,.的区别。
1. 定义 2. 对比
那么[1]有什么区别呢?
2.1.
当我们比较一个read时,除了要问它在基因组中可能的位置外,还要问核苷酸和核苷酸之间的确切对应关系。 例如,我们想要得到类似于“ 可能起源于 chr1 位置 123 到 140。前 7 个核苷酸是 foo 和参考之间的完全匹配,然后是 3 个核苷酸插入,然后是核核苷酸的其余部分匹配 foo 和参考。”
2.2.
当我们进行阅读时dnastar序列比对dnastar序列比对,我们只是在问,“它是从哪里来的?” 不过,我们不一定关心读取与其来源之间的对齐。
直到最近,它们几乎是同义词。 工具喜欢并改变它,因为它们可以在不查看精确比对的情况下将读取分配给基因/特征/任何东西。 因为这样更快,而且我们实际上并不关心一般的对齐结果,所以这在个别应用程序中是一个巨大的优势。
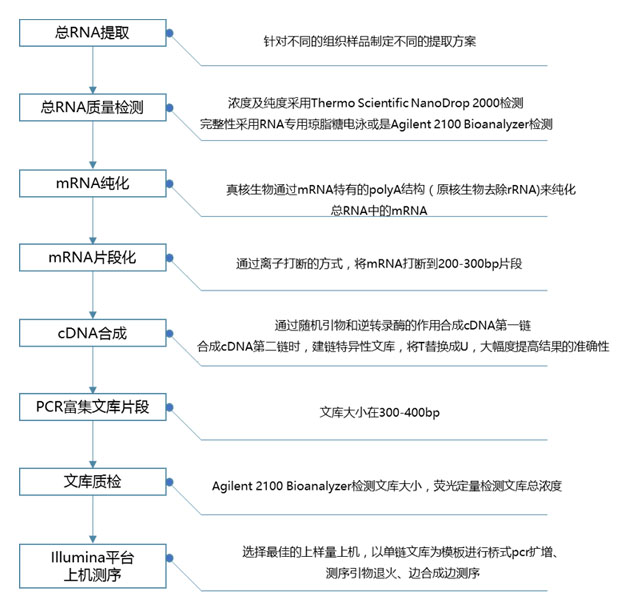
3. 区别 3.1. 星星
STAR[2] 是一个。 它的工作是找出每个测序读数在基因组中的位置,这就是(主要)它输出的内容 - 读数列表及其在基因组上的坐标(以 BAM 文件的形式)。 它非常关心每个核苷酸的正确位置。
对于表达分析,您需要将这些位置转换为基因表达值。 有几种方法可以做到这一点,但最简单的方法是估计基因组中与每个基因重叠的位置的读数数量(执行此操作的程序示例是 )。 另一种方法是使用统计模型将读数分配给可能的转录本(其中多个转录本可能在基因组中重叠)。 执行此操作的程序示例是 RSEM。
3.2.&
并且是定量工具——它们获取包含测序读数的文件并输出基因表达水平。 事实上,弄清楚这些读数在转录组中的来源是该过程的第一步。 一旦找到,他们使用统计模型将其转换为转录表达水平,考虑读取来自哪些转录本。
看起来和做的更多,所以他们应该花更长的时间,但事实并非如此——事实证明他们要快得多。 这一代工具最初是基于这样的想法,即如果你正在量化基因,你更关心的是 read 可能来自的转录本集,而不是它在转录本/基因组中的精确位置。 他们以不同的形式来做到这一点——使用“伪对齐”,最新版本使用一种叫做“选择性对齐”的东西,它介于 STAR 和(据我所知)之间,尽管旧版本被称为准对齐。
4.异同 与单独使用STAR+量化相比,求和速度更快,占用显存更少。 它们提供转录水平的表达信息(其中 STAR+ 计数仅提供基因水平,而 STAR+RSEM 提供转录)。 处理读取映射到多个转录本或基因的情况。 一般来说,只有转录组(由细胞形成的转录物序列)而不是基因组被描绘出来。参考文献
[1]:#
[2]:#
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码