
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-08-12
浏览次数:0
用于基因组学、转录组学和临床研究的综合 NGS 软件
在基因组学和转录组学领域独一无二。 该软件由我们革命性的、易于使用的汇编程序提供支持,使您能够在短短几分钟内建立复杂的基因组测序项目,并手动执行通常需要在其他软件包中进行大量自动干预的任务。
直观的项目设置和分析,与我们获得专利的组装算法相结合,还使您能够以无与伦比的轻松和速度组装和对齐 NGS 数据,以便您可以专注于结果。 不再需要在软件工具之间切换来组装序列、识别重要变异和识别差异表达基因。 你需要的一切都在这里。
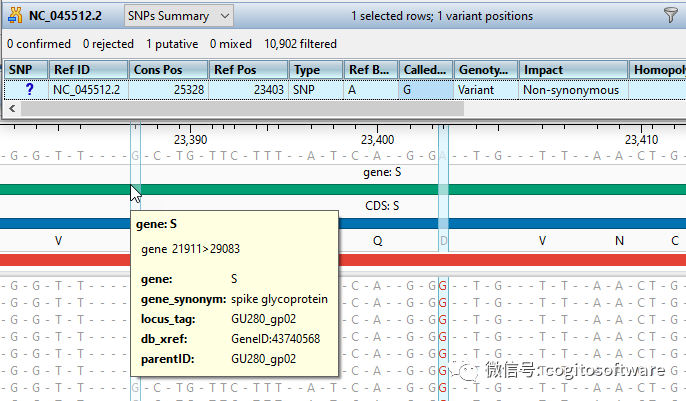
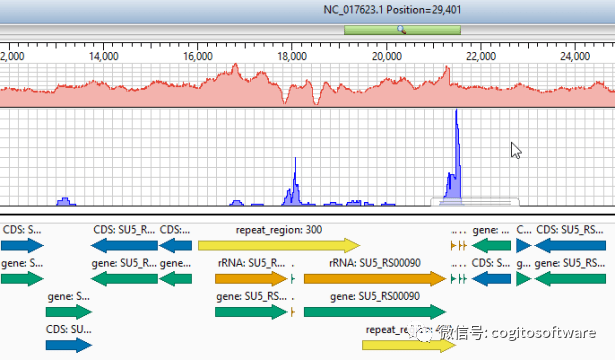
工作过程
ChIP-Seq 数据分析
用于轻松组装 ChIP-Seq 数据并执行峰值测量,使您能够更快地执行全面的 ChIP-Seq 数据分析。
ChIP-Seq 数据分析通常是一个漫长的过程,需要复杂的项目设置以及耗时且复杂的数据操作。 易于使用的向导可在几分钟内指导您完成项目设置,使您能够快速轻松地设置 ChIP-Seq 分析项目。 该软件还包括多种不同的可定制选项,用于变异检查、标准化和峰值测量。 通过使用输入模式或使用位置权重矩阵来识别结合位点,从转录因子数据库中选择结合蛋白。 ChIP-Seq 数据组装后,使用实时交互式视图和全面的过滤工具轻松剖析峰值附近的基因。 基因本体论用于识别具有特定生物学功能的基因之间的关系或确定基因在特定过程中的相对重要性。 可视化峰以解释基因表达和调控,并创建高质量、高度可定制的图像以供出版。 使用正在使用的 ChIP-Seq 分析工具更快地获得全面的结果!
临床研究
使用人类基因组分析手动工作流程在几分钟内构建您的临床研究项目。
近年来,随着对三重分析、肿瘤与正常配对比较和罕见变异研究等工作流程的需求下降,临床研究软件越来越受欢迎。 然而,大多数用于分析人类基因组的商业软件需要大量的前期工作来规划组装和最终的分析项目。 它在临床研究软件领域脱颖而出的部分原因是我们革命性的向导,使您能够在短短几分钟内建立整个基因组或外显子组重测序项目。 手动执行通常需要大量自动化干预的任务,包括手动组织实验和重复、内置访问人类和模型生物的多个基因组模板包、自动合并 BED 和 VCF 文件以及手动检查、注释和分析的变体。 一旦组装完毕,可用的人类基因组分析工具可以立即开始广泛的多样本变异比较,识别统计上显着的变异,并搜索与感兴趣的变异交叉的相关基因。
基因组从头组装
使用 轻松准确地从头组装基因组。
从头基因组组装是一种从大量 DNA 片段构建基因组的方法,而无需事先了解此类片段的正确序列或顺序。 这可能是一个非常困难的估计问题,但由于序列数量庞大、重复区域的存在以及所需的大量 RAM,通常存在速度和准确性问题。
通过指导您通过各种方式满足您的需求,使从头开始组装变得容易。 在我们革命性的基因组组装软件的支持下,用户可以在短短几分钟内建立复杂的测序项目,任务自动化功能可以以无与伦比的轻松和速度驱动从头序列组装,即使在配备适度的计算机上也是如此。
除了传统的从头工作流程(通常对于配对或双端数据最有用)之外,各种基因组整理工作流程可用于基因组草图的纠错和细化。 我们还提供多种工作流程(目前处于测试阶段),用于从头组装和建立来自(包括读长)的长读长测序数据。
无论您采用哪种方式,它都会创建尽可能精确和完整的装配体,为您提供每个完全可编辑装配体的详细统计数据,以及卓越的可视化和装配后分析工具。
从头转录组组装
使用持续的手动工作流程简化从头转录组组装。
从头转录组组装是非模型生物研究中的一项强大技术,但由于数据量巨大以及去除污染物、识别和注释转录本以及使用组装转录本方面的常见困难,转录组数据分析通常会带来挑战。 最大的挑战在于下游分析。 的转录组组装工具。 我们获得专利的降维和组装算法可以在台式计算机上处理几乎任何大小的数据集,从而无需超级计算集群。 手动清理最常见的适配器以及任何用户指定的污染物序列,并使用来自 的转录本注释提供对我们专有转录本注释数据库的访问,从而促进组装和注释。 一旦从头转录组组装完成,我们将提供已识别的新转录本的 FASTA 文件,这些文件经过完整注释并准备好进行下游分析,包括用作 RNA-Seq 分析的参考集。
宏基因组组装
使用强大的算法将宏基因组序列数据与不同的参考数据集进行比较,或降低新序列的维数以进行识别。
考虑到所涉及的数据量,以及随着时间的推移通常需要许多步骤来识别变异、计算覆盖度和消除宿主 DNA,有效组装和解析宏基因组测序数据可能具有挑战性。 通过提供向导来指导您从头到尾完成宏基因组组装和宿主去除过程,从而简化流程。 我们的微生物组分析软件可以轻松地将宏基因组测序数据与来自 NCBI 的数千个完整注释的真菌基因组或数据库进行比对。 恩智浦强大的算法以前所未有的速度执行宏基因组组装,使您能够更快地从原始数据转向分析。 还可以对新序列进行从头宏基因组组装,或对来自 HLA、抗体区域、病毒群体或线粒体群体的高度可变序列进行从头组装。 在宏基因组组装过程中,变异检查和覆盖度估计是手动为您完成的,因此组装后,您可以解剖重叠群并轻松识别微生物组中哪些真菌物种最丰富。
RNA-Seq 比对和分析
使用 RNA-Seq 数据的完整视图。
RNA-seq 是目前探索转录组的领先技术之一dnastar序列比对,转录组对于连接基因组信息及其功能蛋白表达至关重要。 但剖析 RNA-Seq 数据已被证明对许多研究人员来说是一个挑战,因为该技术的应用非常广泛。 快速轻松地将任何大小(从真菌到人类)的 RNA-Seq 或 miRNA 数据集与参考基因组进行比对,然后对比对数据进行广泛的剖析,以便您可以深入研究。 使用 EdgeR 或对差异基因表达进行统计分析,使用全面的过滤工具轻松识别感兴趣的基因,使用基因本体论识别具有已知生物功能的基因之间的关系,使用图表轻松可视化和剖析 mRNA 异构体,查看测序中测量的变体当我们开始 RNA-Seq 分析时,我们并不总是知道我们在寻找什么。 让您可以轻松探索兴趣点,从而获得数据集的概览。
变异分析
用于变异调用和分析的手动软件
从 DNA 测序到解释遗传变异的过程可能漫长而复杂。 借助无数的软件工具和管道dnastar序列比对,剖析原始序列数据的变异可能需要掌握多达十几个生物信息学应用程序和在线数据库。 过程中的每一个额外步骤也会影响结果的确定性和完整性。
Step In,完全集成的变体分析和注释管道,具有直观、易于使用的界面。 其变异注释数据库极大地简化了变异分析,使您无需生物信息学博士学位即可获得明确的答案。
使用精确识别多个数据集之间的显着变化
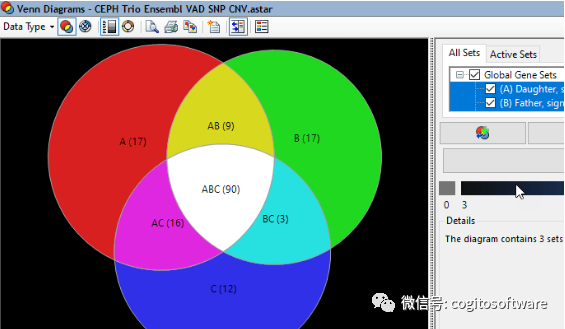
是一个手动管道,可将您的读数组装到模板中,执行变体调用,然后比较多个 NGS 或长读数据集的变体,所有这些都无需人工干预。
事实证明,我们的变体调用工具比我们的商业和开源竞争对手更准确,因此您可以放心,您可以信任结果。
借助强大的过滤工具、丰富的图形视图以及对小型变异数据库(包括 dbSNP、GERP 和 . 它甚至可以让您轻松比较和分析来自其他 NGS 软件管道的多个 VCF 文件,并使用我们的自定义基因组模板包中的信息对它们进行注释,以进行丰富的变异分析。
变异分析软件功能
病毒基因组分析
快速鉴定新病毒株。
从原始病毒基因组测序数据到最终鉴定新毒株可能是一个劳动密集型过程。 对于快速传播的病毒,快速剖析数据的能力至关重要。 我们的病毒基因组测序分析工作流程可用于 NGS 数据,还包括对使用 ARTIC 网络合同生成的 PCR 扩增片段的全面支持。 我们简化了这个有时很复杂的过程,以便您可以更快地进入分析阶段。 首先组装病毒基因组测序数据以评估变异并为每个样本生成共有序列,然后快速将大量基因组与已知弧菌物种进行比对,以轻松识别和比较比对中的变异。
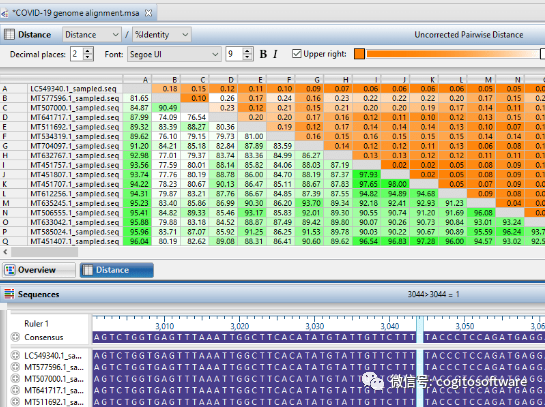
全外显子组/全基因组测序
摆脱全基因组和全外显子组测序分析的艰苦工作,以便您可以专注于结果。
全外显子组测序和全基因组测序产生的海量数据给许多研究人员带来了组装和剖析数据的挑战。 无论您处理的是全基因组、全外显子组还是其他靶向测序数据,所产生的大量信息都需要大量的数据处理和分析。 它还使您能够以无与伦比的轻松和速度将所有主要 NGS 平台的重测序数据与参考序列进行比对。 全面的组装后分析选项可以轻松识别和比较遗传变异以及结构和非编码变异。 需要先进的基因过滤来确定突变对每个基因的破坏程度。 旨在处理大量数据 - 让我们来完成艰苦的工作,以便您可以专注于结果。
应用
基因组序列组装
使您能够在几分钟内构建复杂的基因组测序项目,然后以无与伦比的轻松和速度组装 NGS 和长读长测序数据。
基因表达和变异分析
先进的过滤工具和丰富的可视化功能使您可以轻松评估基因表达并识别多个样本中的显着变异。
序列组装分析与编辑
提供组装后分析工具,例如评估覆盖范围、分析比对背景下的变体以及重叠群编辑。
基因组可视化软件
使用高度可定制的图表可视化基因组测序结果,例如覆盖度和峰值图,这些图表可以轻松导入以进行协作或出版。
*
基于云的存储和组件
提供 NGS 数据管理。 * 将您的 NGS 数据与我们的云估算资源对齐和组合,从而释放您的本地计算机来执行其他操作。
比较套餐
(最受欢迎)
包含的应用程序
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
(每个 Nova 应用+1 次预测)
✔
✔
✔
✔
✔
✔
结尾
公司名称:深圳市者翔软件有限公司
上海哲学官方网站:.com
上海哲学陌陌公众平台账号:
上海哲学软件微博:哲学软件
上海哲学软件邮箱:sales@.com
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码