
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2024-01-11
浏览次数:0

阅读时间:全文共3节,约6400字,约11分钟
获得人类基因组全外显子组离线数据的fastq文件后,如何进行后续的突变检测? 首先要做的是将测序获得的读数与人类基因组参考序列进行比对。
人类基因组参考序列是如何获得的?
随着人类基因组计划(Human,HGP)的进展,Human于2001年首次公布了人类基因组序列草案。2003年,人类基因组计划宣布完成。 HGP样本的来源是大量匿名欧洲捐献者(采集血液或精子),然后选择少量样本提取DNA。 为了保护捐赠者的身份,研究人员并不知道随后对谁的DNA进行了测序(PMID:)。
2004年发布了更准确的基因组序列,版本号是NCBI Human Bulid 35,2004年发表的一篇文章(PMID:)详细描述了该版本基因组的组装过程。 Bulid 35版本的基因组序列包括2.85个核酸,只有341个缺口(缺口主要是由重复片段造成),覆盖了99%的基因组序列。
随后,GRC()在2009年基于NCBI Human Bulid 35版本发布了(hg19)版本,总长度为3,137,144,693; 并于2013年发布了一个版本,总长度为3,209,286,105。
目前,很多关于人类基因组的数据库,如dbSNP等数据库,都已经更新到最新版本。 至于具体分析过程中使用哪个版本的参考基因组序列,您可以根据自己的需求进行选择。
各版本的人类基因组详细信息请参见:
各版本详细下载地址请参见:
#人类)
如何使用BWA进行序列比对?
人类基因组参考序列的来源、详细信息和下载地址都已经知道了,那么我们看看用什么软件或算法将测序数据比对到31亿碱基序列呢?
目前使用最广泛的软件非BWA莫属。 BWA软件除了最常见的平台测序数据外,还可以用于SOLiD、454、reads、reads等。
Heng Li发布的BWA(-)软件目前包含三种算法,分别是2009 BWA-算法(BWA-ALN;PMID:)、2010 BWA-SW算法(PMID:)和2013 BWA-MEM算法(arXiv: 1303.[q-bio.GN]):
1)BWA-ALN算法可用于读长小于或等于100 bp的测序数据;
2)BWA-SW算法可用于读长在70 bp到1 Mb之间的测序数据;
3)BWA-MEM算法可用于读长在70 bp到1 Mb之间的测序数据; 与BWA-SW相比,该算法更快、更准确; 与BWA-ALN算法相比,它可以比较70 bp和100 bp之间的reads。 ,该算法也具有较好的性能。
在使用这三种算法之前,需要为参考序列构建FM索引(全文索引空间)。 FM-index是一种基于全文压缩和索引构建的算法。
BWA 指数 hg19.fa
建立FM-index后,您可以选择三种算法之一进行比较:
-end --- aln算法:
bwa aln hg19.fa 读取.fastq> .sai
bwa samse hg19.fa .sai 读取.fastq > aln.sam
-结束---bwasw算法:
bwa bwasw hg19.fa .fq > aln.sam
-结束---内存算法:
bwa mem hg19.fa 读取.fastq> aln.sam
-end --- aln算法:
bwa aln hg19.fa .fq> aln1.sai
bwa aln hg19.fa .fq> aln2.sai
bwa 样本 hg19.fa aln1.sai aln2.sai .fq .fq > aln.sam
-结束---bwasw算法:
bwa bwasw hg19.fa .fq .fa> aln.sam
-结束---内存算法:
bwa mem hg19.fa read1.fq read2.fq > aln.sam
比较后得到的信息存储在SAM文件中。 尽管 SAM 文件只有 11 列必填字段,但它包含的信息量非常丰富。 接下来我们看一下该文件中存储了哪些信息。
SAM文件格式简介
SAM文件格式是Heng Li在2009年提出的用于存储比较结果信息的文本文件(用Tab键分隔),同时他发布了处理SAM文件的软件(PMID:)。
SAM文件包括两部分:注释信息( )和比较结果部分( )。
注释信息
这里主要介绍三个信息:@SQ、@PG、@HD。
@SQ
参考序列的描述
示例:@SQ SN:chr1 LN:
SN:人类参考基因组中的染色体编号,如chr1、chr2
LN:参考序列中序列的长度
@PG
所用程序的描述
示例: @PG ID:bwa VN:0.7.13-r1126 CL: bwa mem hg19.fa read1.fq read2.fq
ID:软件名称
VN:软件版本号
CL:命令行
@高清
使用 SO 记录对齐读取的顺序。
例子:
所以:
所以:
所以:
或者sam文件中没有@HD这行信息:比较后的默认排序顺序与比较时输入的fastq文件一致。
那么如何对比较后的文件进行排序呢? 使用软件中的排序模块。 该模块需要输入bam格式:
查看 -Sb aln.sam > aln.bam
下图展示了不同排序方式下的sam文件:
我们先看一下fastq文件中的排序顺序
fastq 文件:less -SN read1.fq

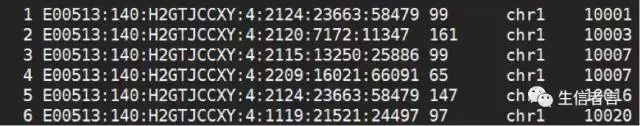
对比后默认的sam文件与对比时输入的fastq文件一致,如下图:
-X aln.bam | 少-SN
注意:可以看到下面文件的第二列是一个字符串,与下面的截图不一致。 这是因为查看时添加了-X参数,可以将第二列中的FLAG转换为字符形式。 FLAG会在比较结果的解释中指定,这里我们主要看FLAG一栏的最后一个数字。 “1”代表末端测序中的read1,“2”代表末端测序中的read2。
按查询名称排序的SAM文件,按(即比较结果部分的第一列)从小到大排序:sort -n aln.bam aln。
将排序后的sam文件按与参考序列对齐的位置(即对齐结果部分的第三列和第四列)从小到大排序:sort aln.bam aln。
综上所述,注释部分主要记录了比较reads时使用的参考序列的基本信息、SAM文件使用的程序以及reads的排序规则。
接下来我们详细解释一下SAM文件中的比较结果部分。
比较结果
比对结果部分中的每一行代表一条read的比对信息(如果是双端测序,则每行记录单端reads的比对情况,该read在SAM/BAM文件中称为Query),包括11个必填字段和其他可选字段由 Tab 键分隔。 共有 11 个必填字段,按固定顺序排列,详细信息如下:
上校
场地
类型
简短的
例子
名称
读入fastq文件
:140::4:1223:25723:72631
旗帜
INT
FLAG 数(请参阅下面的详细信息)
聚丙稀
名称
此读数与参考序列中的哪条染色体进行比对?
chr1
销售点
INT
此read比对到染色体上的位置,并且是从1开始的,即参考序列的第一个碱基位置编号为1(从0开始,参考序列的第一个碱基位置编号为0)
13198
MAPQ
INT
就是Phred-:MapQ = -10 log10(P),比如MapQ=30,表示匹配到这个位置的概率是千分之一。 与MapQ=20相比,不是随机事件,reads比较更准确。 精确的。
48
雪茄
简要对比信息表达(详见下文)
150M
近端串扰
示例中,末端测序的read为read1,信息为与其配对的read2相比的染色体编号(如果末端测序的read为read2,则信息为与其配对的read1编号相比的染色体编号):“= " 表示与读到的一致; 如果没有,则填写相应的染色体编号。
PNEXT
INT
另一个读数在染色体上的位置与末端测序中的该读数配对。
13019
特伦
INT
根据 RNAME+POS 和 RNEXT+PNEXT 计算这对 read 对应的 DNA 文库片段长度,如下例所示: POS – RNEXT + CIGAR 中的 M+I 编号 = 13198-13019+150 = 329。则为什么是负329? 因为read1是在read2的下游对齐的。
-329
10
序列号
该读段的碱基序列
……
11
质量
read碱基序列对应的质量值
第二列是FLAG,FLAG是标识符的总和,各种比较情况用不同的数字表示,每个数字代表一种比较情况。 将符合一定比较条件的查询编号相加dnastar序列比对,得到的编号就是FLAG。 FLAG 详细信息如下表所示:
标志(十六进制)
标志(十进制)
通过末端测序获得的单端读数
该标志仅在 0x01 存在时才有意义:末端测序中的配对 read1 和 read2 与参考基因组上的适当位置对齐。 详细解释见下图1: 上图---read1和read2对对齐到同一条染色体上; 下图---read1和read2对齐到不同的染色体。
详细说明1:
该读取本身并未映射到参考基因组dnastar序列比对,请参见下面解释 2 中的插图。
该Flag仅在0x01存在时才有意义:与其配对的read没有映射到参考基因组,参见下面详细解释2中的图示。
详细解释2:
16
read本身与参考基因组的负链进行比对,SAM文件中第10列的SEQ是fastq文件中碱基序列的反向互补。 详细说明见3。
32
该标志仅在 0x01 存在时才有意义:与其配对的读取与参考基因组的负链对齐。
详细解释3:
:140::4:1101:11475:2381 的 read2 与参考基因组的负链对齐。 原始fastq文件中的序列是:
。 。 。 。 。 。
在SAM文件中,第十列SEQ为:
。 。 。 。 。 。
64
该标志仅当 0x01 存在时才有意义: -end read1
128
该标志仅当 0x01 存在时才有意义: -end read2
256
该比对信息不是read的最佳比对位置。 请参见解释 4 中的插图。
详细说明4:
第512章
读取失败/
1024
该read是由PCR或()引起的read,即至少有一个其他read的碱基序列与该read一致。 PCR过程是DNA模板的复制。 如果测序数据量足够大,就有可能检测到具有相同碱基序列的reads。 光学重复简单理解就是信号本身来自于测序、拍照获取信号过程中的信号。 ,但被识别为两个,导致出现具有相同碱基序列的两个读数。
FLAG如此之多,如何快速提取特定FLAG的reads呢? 只需在查看后添加-f参数即可。 例如,要提取未映射到参考基因组的读数:
-f参数指定FLAG对应的十六进制值:查看-X -f aln.bam | 少-SN
-f参数指定FLAG对应的十进制值:view -X -f 4 aln.bam | 少-SN
第六列CIGAR是一个简短的比对信息表达式( ),它基于参考序列,用数字加字母表示比对结果,其中M-Match/; 我-; D-; S-软夹;H-硬夹; P-; N-从 ,例子如下:

综上所述,对于末端测序来说,SAM文件的比对结果部分中的每条信息不仅详细说明了本次read的比较情况(如FLAG、RNAME、POS、CIGAR),还记录了其匹配reads的比较情况。 情况(例如 FLAG、RNEXT、PNEXT)。
您对比对过程中涉及的人类基因组参考序列的来源、比对软件、比对结果文件SAM/BAM是否有了更深入的了解呢?
下一期我们来谈谈如何进行突变检测。
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码