
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2024-01-24
浏览次数:0
1 背景知识介绍
进行 PCR 反应时需要寡核苷酸引物 (rs)。 我们需要设计与 DNA/cDNA 模板区域互补的引物。 当一次添加一个核苷酸时,核苷酸上的反应基团 ( ) 必须选择性地封闭 ( ) 和解除封闭 ( )。 引物的主要特点是它们必须与模板分子上的序列相对应(必须与模板链互补( ))。
当然,引物不需要与模板链完全对应(例如添加酶切位点或构建突变位点的引物); 但引物的3'端必须与模板DNA链完全对应(所以当我们添加酶切位点时,它是5'端),这样才能延伸。 通常3'端使用鸟嘌呤(,G)或胞嘧啶(,C),尽量不要有腺嘌呤(,A),引物的5'端通常有几个核苷酸延伸()。 另外,杂交引物( )的3'端必须相互指向,因此我们在合成引物时,总是从5'端开始向3'端开始。
引物的长度对于扩增特定序列很重要。 短引物主要用于扩增小而简单的DNA片段。 另一方面,长引物用于扩增真核基因组 DNA 样本。 但是,引物不应太长(>30bp 引物)或太短(<15bp)。 当然,当我们的模板很纯的时候,引物就不受这些规则的限制,可以根据实际情况定义引物的长度。 短引物更不准确。 也就是说,非特异性DNA扩增产物增多,表现为电泳时出现很多非特异性条带。 长引物的缺点是它们导致杂交速率较慢。 平均而言,待扩增的 DNA 片段大小应在 1-10kb 范围内。 实践告诉我们,我们比较放大的长度是3Kb及以下,当然这也取决于物种。
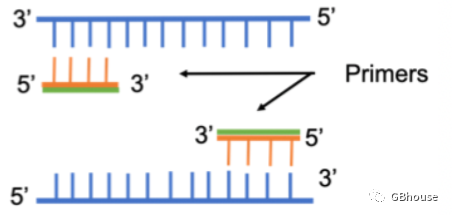
(图1引物扩增示意图。PCR基因扩增方向与体内合成方向一致,即从5'端到3'端。)
引物设计还应注意以下规则:引物的结构应相对简单,无内部二级结构,避免内部折叠; 我们还需要避免引物-引物退火,这会产生引物二聚体并破坏扩增过程; 设计时如果不确定引物的某个位置放置什么核苷酸,可以在该位置包含多个核苷酸,这称为混合位点; 您还可以使用基于核苷酸的分子插入物(肌苷)来取代传统的核苷酸,以获得更广泛的配对能力。
考虑到上述信息,底漆通常应具有以下特性:
(1)长度为18-24个碱基;
(2)G/C含量40-60%;
(3)以1-2对G/C开始和结束;
(4)退火温度(Tm)50-60℃;
(5) 引物对之间的Tm差异应在5℃以内;
(6)引物对不应有互补区域;
(当然这是在比较理想的状态下,如果状态不理想,可以尝试以此作为参考)
2 NCBI引物设计实践
2.1 定量引物规则
最重要的是特异性(即只扩增需要检测的基因)。 其他规则(1)引物长度为20-21 bp; (2)避免引物中连续出现5个或更多相同碱基,如AAAAA或GGGGG; (3)每个引物两端的碱基最好是G或C(GC碱基之间有3个氢键,以保证引物与模板连接的稳定性); (4)GC含量为45%~55%; (5) PCR产物大小为85~300 bp; (6)引物应尽可能跨越内含子和外显子; (7) 参考上节引物设计规则; (八)其他。
2.2 NCBI设计定量引物
2.2.1 以NCBI查mRNA序列(以人TNF-α)为例
(1)寻找人类TNF-α基因。 方法如图2-1所示。 点击
(图2-1 NCBI检索人TNF-α基因)
(2) NCBI定位需要引物设计的基因并左键点击。
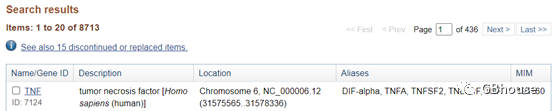
(图2-2 选择我们需要研究的TNF-α)
(3)TNF-α相关信息()
因为我们只是设计引物,所以不需要阅读太多的信息。 但值得注意的是,有些基因有很多剪接体,因此选择合适的剪接体非常重要。 如果我们知道具体研究哪个拼接体,直接选择即可; 如果我们不知道要研究哪个剪切体,只需选择最长或研究最多的剪切体即可。
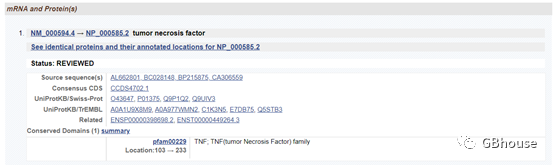
(图2-3 TNF-α剪接体信息,共包含1条信息,用于qPCR引物设计)
(4)点击:.4进入mRNA详细信息页面()
A。 查看信息,包括每个外显子、内含子和 CDS 区域

(图2-4查看外显子、内含子和CDS区域信息,因为设计是为了跨越外显子-内含子)
b. 第一个外显子在1-363之间,CDS区域在178-897之间。 让我们学习一下定量引物设计的规则。 一般定量引物会选择mRNA的前500bp或者第一个外显子或者CDS,所以这里我们选择TNF-α的前600bp(当然具体长度可以适当改变)用于定量引物设计(序列可以复制或另存为文件)。
(5) NCBI引物设计
这里我们使用NCBI的BLAST功能选项进行设计
(a) 打开NCBI网站()dnastar引物设计,找到BLAST选择框,如下图红框所示;
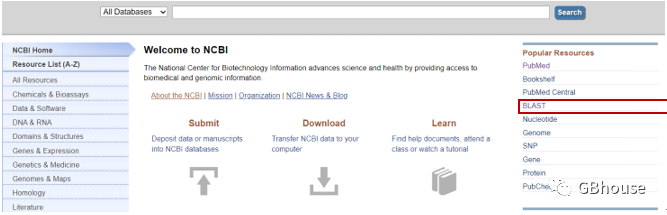
(b)找到“-BLAST”选择框并左键单击
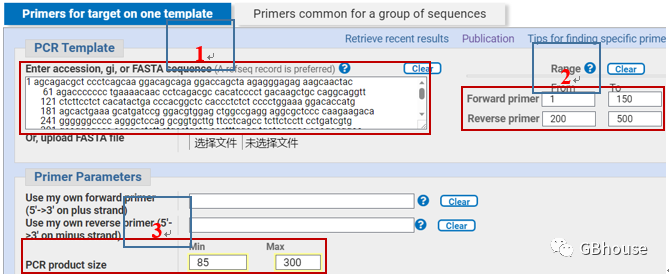
(c) 新页面优先:输入基本序列,或从文件中拖动序列; 然后限制上下游引物的起始位置(记住qPCR产物在85-300bp之间的约定,其他规则软件会自动设置(固定的,不用担心),这里我们选择1-150的位置设置为上游( )-200-500 下游( );产品长度设置为85-300,如下图所示。
(d) 点击本页底部的“获取”;
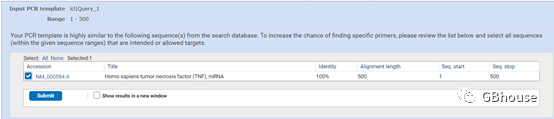
(e) 检查并提交序列所属基因信息,勾选复选框并点击“”;
(f)等待界面,软件设计引物需要时间,几分钟甚至更长时间,等待即可……;
(g) 获取引物信息。 这里需要检查引物的特异性,检查引物下面是否还有其他mRNA信息。 如果不包含,说明该引物特异性极高,只能扩增我们需要的mRNA(TNF-α)。
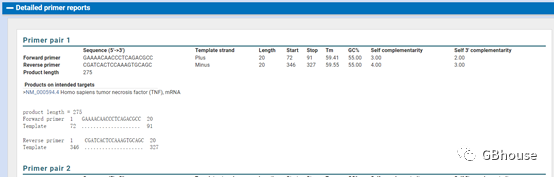
(h) 保存所需的qRT-PCR引物(当引物较多时,以最上面的引物为主,为了使实验尽可能成功,建议合成两对或更多对定量引物)
:
:
7定量引物设计实践
俗话说,有太多的技能需要克服。 这个概念对于设计引物也是一样的。 在极少数情况下,NCBI 设计的引物可能无法用于实验。 例如,熔解曲线不是标准峰(非单峰),不能靶向基因。 靶向效率低等。 因此,为了实验的成功,掌握另一种设计定量引物的方法是极其有必要的。 对了,文末附上了oligo 7的下载位置,不然你就说我流氓了!
3.1 寡核苷酸引物设计规则
具体规则如上所述。 只要回头看看。 如果你还不知道如何露手或露屁股dnastar引物设计,我就打你三巴掌,让你再做一次。
3.2 使用oligo 7设计定量引物
(a) 打开oligo 7软件
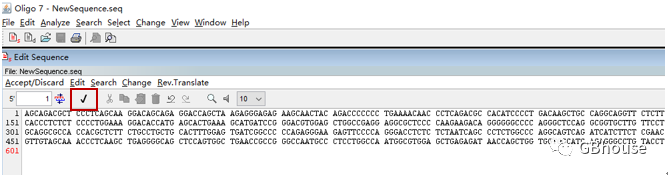
(b) 点击:file-new,然后将序列粘贴进去,点击红框中的“√”;
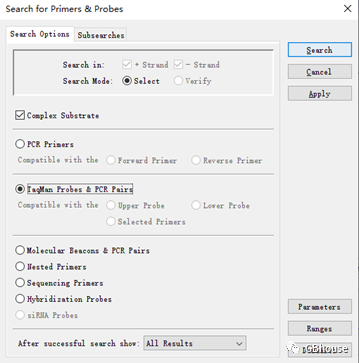
(c) 点击“- for & probs...”,然后选择“probs & ”/当然,你也可以选择“PCR”,但是会很多,所以点击“”即可;
(d) 获取引物信息
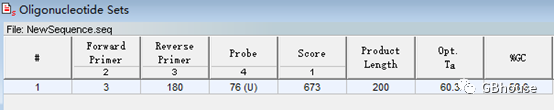
e) 保存定量引物,一定要选择要保存的引物,“文件-保存-”,选择位置(如果找不到,别怪我没说清楚)并给它命名;
上游底漆:
下游引物:
(f) 合成引物(导出的引物都是5'端到3'端,直接发送合成即可):
:交流电
:CTC
(对了,导出的引物序列需要软件打开,最后也发到这里(),下载地址:;提取码:1179)
(写在最后,欢迎转载引用传播科学价值!!!关注博主,有用的话点赞+讨论是博主继续更新的动力!!!)
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码