
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2025-05-24
浏览次数:0
1 背景知识
1.1 基因过表达简介
在科研探索中,采用经典遗传学方法,往往以对特定表型变异的研究为起点。得益于现代高效测序技术的助力,研究者能够迅速获取有关某特定表型中基因、蛋白质、离子等表达情况的信息,进而验证特定基因在表型形成过程中的作用。
关于基因,人们所实施的一系列操作通常被称作基因工程。然而,随着时代的变迁,“基因工程”这一概念已经超越了其最初的生物学范畴,获得了更为丰富、更为广泛的理解。今天,我们将重点探讨基因工程在生物学领域的价值和作用。在生物学实验室中,我们学习到的基因工程,亦称作遗传工程、转基因或基因修饰,它是一种通过生物技术手段直接对有机体的基因组进行操作,旨在改变细胞遗传物质的方法。在实验或论文汇报中,我们经常接触到几种主要的基因编辑手段,包括基因过表达技术,也称作OE或基因敲入技术,即KI;还有基因敲低技术,简称KD;以及基因敲除技术,标记为KO。
基因剂量在早期阶段对维持正常基因功能至关重要,这一重要迹象可以通过核型分析观察到。它揭示了非整倍体现象,这一现象是导致人类遗传综合征、果蝇及植物突变表型的根本原因。因此,与基因内部离散突变无关,整个染色体或其部分区域的简单拷贝数增加,也可能引发突变表型。基因过表达,即通过体外克隆技术,将目标基因组装至特定的穿梭质粒或病毒载体中,借助载体上构建的调控元件(通常包括强大的启动元件),在人为设定的条件下,促使基因实现大量的转录与翻译,进而达到目的基因的高效表达。
1.2 过表达的常见途径
搭建起适宜的表达平台后,其应用范围十分广泛(如图1所示),包括但不限于:在野生型过表达载体改造的生物体中观察其效应;研究过表达突变蛋白的功能;通过过表达技术探究基因与蛋白之间的互作网络(如基因-蛋白互作、蛋白-蛋白互作等);利用过表达技术验证修饰位点;筛选针对特定靶点的药物;在异种细胞中实现基因的过表达(如在低表达细胞中过表达高表达基因);以及研究过表达带来的上位效应等。
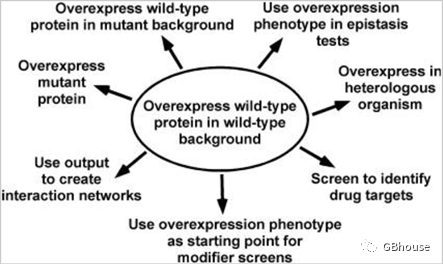
图1 展示了过表达技术的典型应用,(参考文献编号:doi: 10.1534/.111.)
1.3 过表达的功能
经过特定基因的表达,生物的某个表型特征可能会受到抑制,亦或是被激活,这种现象通常符合我们的预期,但有时也可能出现无效果的情况,或者其他不同的结果。
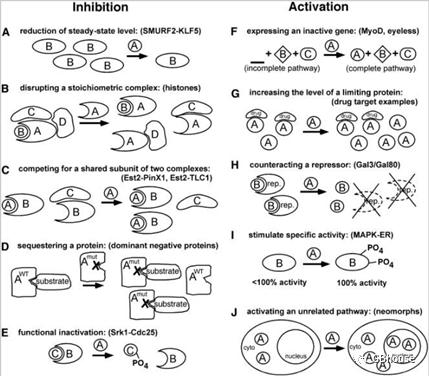
图2展示了基因过表达的作用机制概览。该机制负责调控过表达表型。过表达机制主要分为两大类,一类是抑制(位于左侧),另一类是激活(位于右侧)蛋白质。具体的作用机制将在下文详细阐述。针对每一种变体,括号内提供了一个在文中进一步详细讨论的典型实例。例如,(A)过表达可以通过改变其他蛋白质的转录、翻译或降解速度,从而直接降低其稳态水平。在多蛋白复合物中,亚基A通过过表达产生多个接触,这会引起A-B、A-D或A-C亚基的局部组装,进而降低构成完整功能的A-B-C-D复合物的数量。如果蛋白B同时参与构成两个独立的A-B和A-C复合物,那么非共享的亚基A的过表达能够有效竞争蛋白质B的有限资源,因此会减少具有功能的B-C复合物的总数。突变酶A即便在底物存在的情况下仍能与之结合,其过表达通过传统的显性阴性或反态机制与野生型酶A争夺同一底物。过表达现象甚至能从功能上使蛋白质失活,且这一过程并不依赖于竞争机制。具体来说,一个亚基在翻译后发生的修饰作用破坏了蛋白质之间的相互作用。此外,即便通路的其他部分保持完好,正常沉默的基因在表达时也能激活其所在的通路。在特定基因表达受限的情境下,提升基因表达水平能够提升整体活性,并激发其输出功能。通过多种途径,如抑制因子的分解、翻译后修饰或直接竞争,可以消除阻遏因子的影响,使其失效,从而激活相关途径。以基因A为例,其过表达会促使阻遏因子(rep.)分解,进而释放具有活性的蛋白B。此外,过表达还能提升其他蛋白质的特定活性。最普遍的机制或许是通过翻译后的修饰作用实现,(J)这种修饰作用能够借助新形态效应激活新的途径。参考文献:doi: 10.1534/.111.
1.4 过表达类型
依据基因在生物材料中表达水平的增强程度,可依据其表达持续的时间差异,区分为短暂性高表达类型以及持续性高表达类型。通常情况下,收集生物样品的瞬时过表达过程需在48至72小时内完成,此时基因的过表达水平可增至数百倍,可通过qRT-PCR等检测方法进行验证;而实现稳定过表达,则需要将过表达的基因整合至生物样品的基因组之中,这通常需要借助病毒载体将靶基因导入基因组,随后还需通过特定的筛选手段,以获取能够稳定表达的正性细胞。在筛选过程中,我们需区分病毒感染细胞但目的基因未实现过表达的样本作为阴性对照,以及实现基因过表达的样本作为阳性组。然而,构建过表达载体的方法在这些样本中是相同的,甚至可以说是完全一致的。
1.4 过表达优点
过表达研究提供了几个优点:
这种工具具有多用途特性,能够适应野生及突变两种生物背景,并以多种形式进行应用。
(2)可以识别监管限速步骤;
(3)具有显性作用,因此在二倍体生物中易于进行;
(4)它甚至为冗余基因提供功能链接;
(5)它识别功能丧失基因的互补相互作用。
2 过表达引物设计实操
2.1 确定过表达质粒
我们采用了最为普遍的质粒pEGFP-N1进行实验。
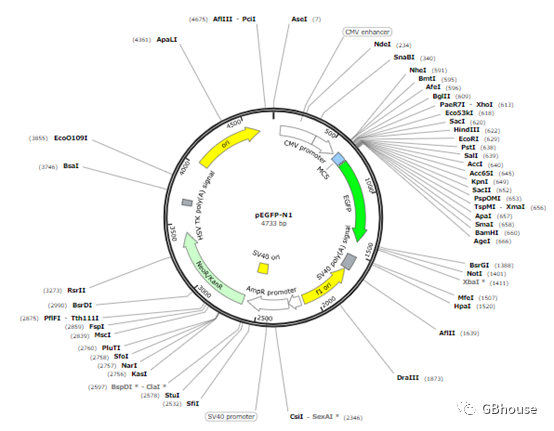
(图3 pEGFP-N1质粒图谱)
质粒图谱解读:
多克隆位点,通常简称为MCS,是指目的基因序列被插入的特定区域。在这一区域中,存在酶切位点,这些位点允许实验者将它们嵌入到目的基因序列中。这一点必须予以充分理解。
绿色荧光增强蛋白,通常简称为EGFP,若将穿梭质粒引入细胞内部,借助显微镜便能够观察到显著的绿色荧光,这一现象可作为目的序列已成功进入靶细胞的验证手段之一。
SV40 poly(A),通常称作猴空泡病毒的PolyA(病毒40 PolyA,亦称Poly A),其功能主要包括两个方面:一是终止基因的转录过程;二是将转录出的mRNA分子添加一个Poly A尾巴(由DNA序列构成,长度约为240碱基对)。mRNA的Poly A结构对于真核生物来说,能够有效保持基因的稳定性,而在原核细胞中,它则能加速mRNA的降解过程。
f1 ori,即f1噬菌体的ori区域,其功能在于调控单链DNA(ssDNA)的复制过程。
ori是质粒在大肠杆菌中的复制起始点,其主要功能是促使质粒DNA以环状方式复制,从而实现质粒在大肠杆菌内的自我复制,这一特性也是选择转化感受态大肠杆菌的重要依据。
氨苄青霉素启动子,即AMP(r),在阳性大肠杆菌(一种原核生物)中具有水解β-内酰胺环的功能,从而消除氨苄的毒性。这一特性在转化涂板过程中至关重要,必须添加,否则整个细菌培养皿将布满菌落。若添加不当,甚至可能完全无法生成菌落。
NeoR/(KanR)中含有的氨基糖苷磷酸转移酶能够使G418(即长那霉素的衍生物)失去活性。通常情况下,仅具备单一抗性的质粒主要是原核克隆载体和原核表达载体。而那些含有两种或更多抗性的质粒,则通常是穿梭质粒。NeoR所代表的真核抗性在这里充当了筛选真核表达过程中阳性细胞的靶点。
HSV的终止子信号以及PolyA结构,共同作用在mRNA上,实现信号的终止,同时PolyA的存在有助于mRNA的稳定性。
生物科技的进步促使众多用途各异的质粒得以研发,这些质粒的信息和功能日益繁复,然而,它们最根本的作用却始终如一。因此,在获得一个新质粒后,务必详尽掌握其相关资料。然而,人的记忆力有限,对于这个质粒,我们需要记住以下关键信息:首先,MCS多克隆位点中两个待插入序列的具体位置;其次,转化至原核菌种所需的抗性,通常以AMPr和Kan r为主;再者,导入真核细胞后可能出现的表型,如绿色荧光等;最后,筛选阳性细胞时可能用到的药物,如G418、嘌呤霉素等。
2.2 获取目的基因序列(以human GPX4为例)
在NCBI数据库中进行搜索,查找关于GPX4的人类相关资料,顺序可以随意。
(b)找到需要的GPX4并单击;
(c)基因简介,初学者可以大概看看;
基因表达在各类组织中的状态各异,需根据所研究的特定组织或细胞类型,对mRNA的浓度进行适当调整(当表达量较低时,可以通过提升cDNA的浓度),从而实现目的基因的快速扩增。
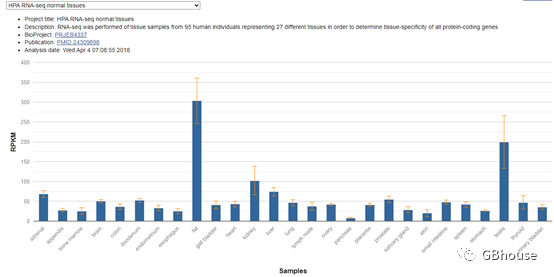
在剪切体信息查看界面,我们能够观察到human GPX4目前存在四个不同的剪切体。依据具体的研究需求,如课题要求、剪切体长度以及研究频率等因素,挑选出适宜的剪切体进行深入探究。在此情形下,我们选取了第一个剪切体作为案例,并点击了编号为.3的选项。
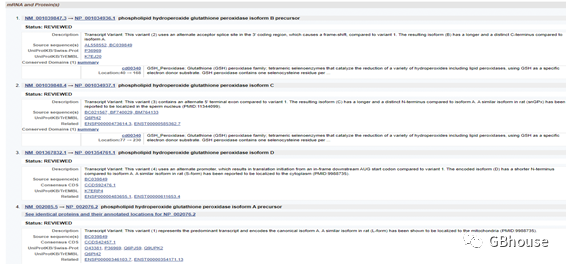
(f)在中定位CDS( )(单击CDS即可);

(g)保存基因序列
1,保存序列
将序列分别选取并复制,然后将它们一同粘贴进txt文档里,即使序列前方的数字一同被复制下来也无妨。
将.txt文件的扩展名更改为.seq,更改后的文件类型将变为可读取状态(具体可打开的软件信息请参阅文档末尾)。
对CDS与mRNA的相对位置进行对照分析。启动软件后,注意查看(安装完毕后,该启动位置会显示为“”)。
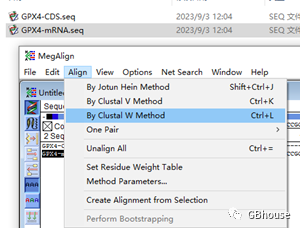
(mRNA和CDS比对方法)
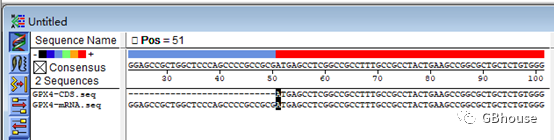
CDS的起始密码子ATG中的A位于mRNA的第51个碱基处。
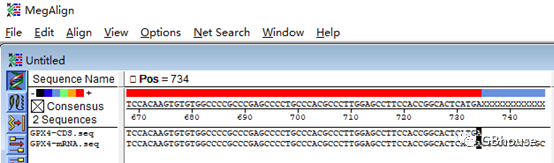
明确CDS的起始和终止密码子TGA位于mRNA的第734个碱基处。
请注意:准确界定起始和终止碱基的位置,这对于在克隆操作中获取一个完整的GPX4编码序列至关重要(若序列不完整,将无法获得有效的序列,更不用说后续将其翻译成蛋白质了)。
2.3 利用oligo 7设计引物
将mRNA序列粘贴入文档:启动新文件创建,操作完成后,点击确认标志“√”。
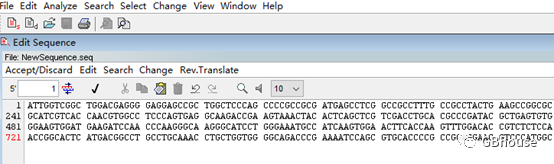
(b)上游引物设计
(定位51号碱基位置)
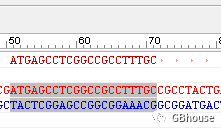
(定义为上游引物: )
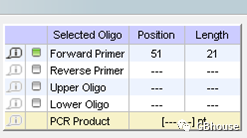
上游引物分析中,需关注引物二聚体的形成情况;同时,发卡结构的稳定性也是关键;此外,还要注意错配位点(site)的存在。
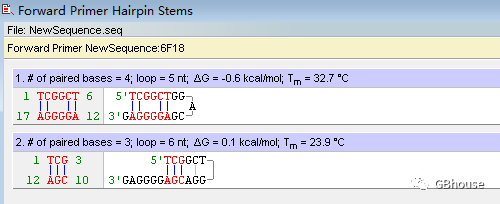
根据分析结果,G值不大于3,所以结论是不符合要求,因此引物序列需要向更前的位置进行调整。

将测序起始点调整至mRNA序列的第6个碱基位置,同时将引物的长度调整为18个碱基长度。

(/ G / ≤ 3,符合要求)
在理想状态下,这一条件通常不应存在,然而在现实情况下,这一目标往往难以实现;然而,若要确保开卡结构的开启,其温度必须严格控制在Tm值以下,这是必须满足的条件。
该网站上的参数XXX(位于上方)越接近底部越好,这一数值对结果影响有限,可能为数十或数百(位于第一行);自第二行起至末尾,评分必须保持在100以下,当然对于设计难度较大的引物,可以适当放宽至150以内。

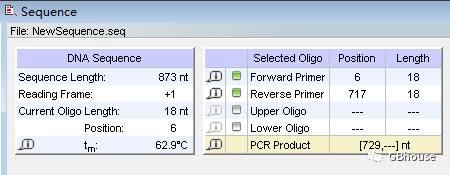
记录了上游引物的温度值为62.9摄氏度,通常情况下,上下游引物的温度差异不会超过3度。
(b)下游引物设计
(定位下游引物的5’端始于CDS的最后一个碱基)
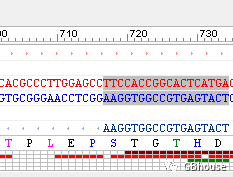
点击图片右侧区域,确保设定下游温度为58.2℃,并注意观察其与上游温度62.9℃之间的差异,若超过3℃,则可适当增加下游引物的长度(即-oligo)。
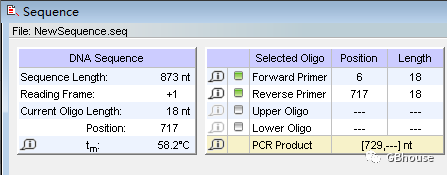
将下游引物的长度增加到20个碱基,其Tm值应为61.6度,以满足上下游引物之间温度差异的标准。需要注意的是,这里的“延长”指的是向引物的末端增加碱基。为此,我们增加了2个碱基。由于这个增加是在引物的末端,我们必须将序列的5'端向前移动2个碱基,以确保下游引物从5'端开始。

对引物二聚现象进行探讨;对发卡结构进行深入研究;对错配位点进行细致分析。
( :符合要求)
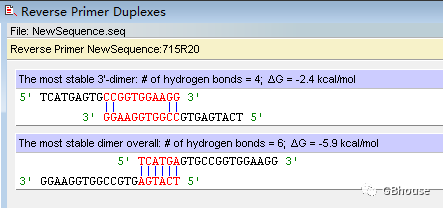
( :符合要求)
该地点:不满足条件,而XXX(位于上方)则符合标准;第二行的高度137虽然超过了100,但接近150,虽可作为备选方案,但在此情况下,我们应选择更佳的选项,并且还需考虑大家如何调整位置以设计下游引物。

调整下游引物的定位(自然地,通常是向后移动1至4个碱基,避免出现连续的4至5个碱基或大量GC序列),使得下游的5'端起始位置位于736处。
接下来,对引物二聚体进行探讨,对发卡结构进行考察,对错配位点进行剖析,此处就不再赘述,分析结果明确指出,下游引物满足所需条件。
(c)保存引物
文件-保存-,根据上下文信息进行分别保存,导出的引物默认设定为从5'端至3'端。
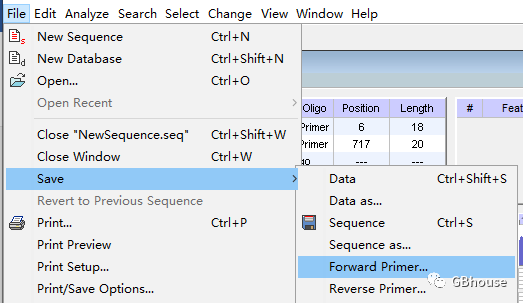
获得的引物序列被命名为F1/R1dnastar lasergene,这主要是出于个人习惯;我倾向于采用两步法克隆CDS区域:首先,我会尽量扩增至目标序列;其次,针对CDS区域,我会再次设计一对引物,并在5'端添加相应的酶切位点。当然,也可以在F1/R1的5'端添加酶切位点。
根据F1/R1设计引物(只需要合成F1/R1即可)
根据图3 质粒图谱的MCS选择
上游:Hind Ⅲ(622) ;
下游:Kpn Ⅰ(649);
上下游的酶切位点最好是相隔2至3个位点,或者更多,尽管市售的质粒可能不需要遵循这一准则,但个人认为保留一两个间隔会更为适宜,这样有助于提升酶切效率,并提高实验成功的几率。
F1:
R1:
依据F2/R2的序列信息设计引物,这一过程需要合成两对引物,分别是基于F1/R1和F2/R2序列的。
在F2/R2的设计中,我们无需再详细阐述相关规则,只需留意并关注上下游的温度变化即可。
上游引物的5'端从ATG位点直接起始,而下游引物的5'端则从TGA位点直接起始,需留意它们的方向是相互对立的。
F2:
R2:
凭借我多年的实践积累,采用设计两对引物的方式,成功的可能性会明显增强。
对了,打开导出的引物序列时需要使用特定的软件,同时,我也将相应的软件一同提供,下载链接已附上(),具体地址为:https://pan.baidu.com/s/pLQ,提取密码是:1179。
在文末dnastar lasergene,诚挚邀请您进行转载与引用,共同推广科学的价值!敬请关注本博主,若内容对您有所帮助,不妨点赞并参与讨论,这将是我持续更新的强大动力!
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码