
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-06-19
浏览次数:0

RNA-seq 是转录组研究的一项基本技术。 自推出以来,已经开发了数百种分析工具,分析中不同步骤所涉及的权衡(如速度、资源消耗、灵敏度、准确性等)是至关重要的。 RNA-seq分析包括序列比对、转录本组装、表达定量、差异分析、可变剪接、融合基因检测、突变分析、RNA编辑等。一般分析不需要贯穿整个过程,可以简化基于你自己的需要。 那么,在给定成本和性能限制的情况下,此类分析工具是否存在确定性的最佳组合?
2017 年,他发表了关于转录组分析过程的研究。 针对15个样本(正常样本、癌细胞和干细胞、短读长和长读长测序数据)的转录组数据,借助39个工具,对约120个常见组合进行了约490个深度分析RNA-seq 的形式,并以测序质量控制联盟 (SEQC) qPCR 测量结果作为阴性对照,以综合评估 RNA-seq 的分析工作流程。 研究人员总结出一套普适过程,如右图所示:
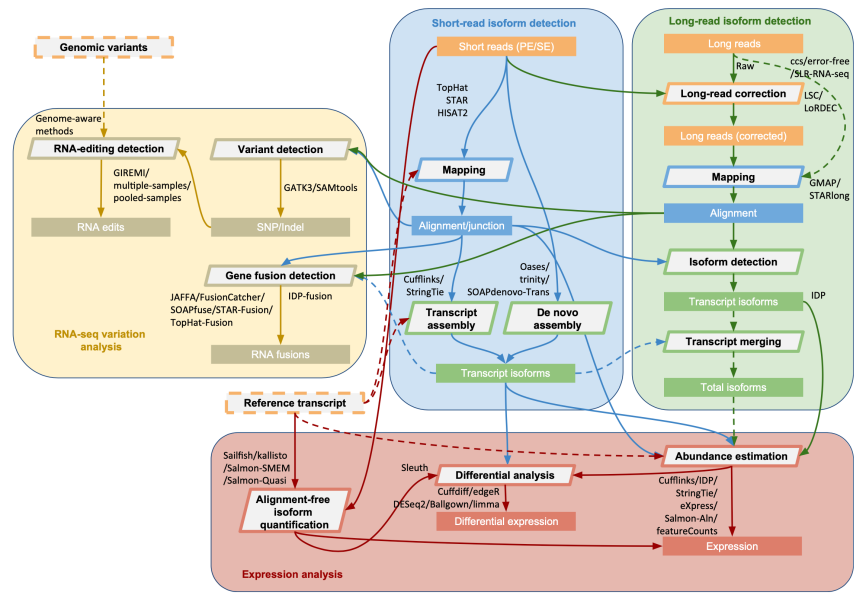
RNA-seq 分析的第一步通常是识别一组表达的转录本,这通常涉及将读数与适当的参考序列进行比对,然后根据比对构建转录本。 一般以基因组或转录组序列为参考dnastar拼接序列,以参考基因组为参考可查新转录本,但需要耗费资源的reads ; reads 使用转录组序列作为参考相对容易,但不允许检查新的转录本。 如果研究物种没有可靠的参考序列(基因组或转录组),可以采用de novo 来鉴定转录本,即没有“参考”的转录组,没有合适的参考序列,组装转录本序列从头开始,然后转录量化书。 下面从几个方面比较不同工具和工具组合的性能。
1. 序列比对质量评估
研究人员评估了 STAR 和 STAR 在对齐和切点预测方面的性能。 STAR 仍然具有最高比例的唯一映射读取对,即在基因组上具有唯一对齐位置的读取比例最高。 与STAR不同的是,STAR只保留-end reads与基因组比对的序列,对低质量比对具有较高的容忍度(容忍更多的错配核苷酸和soft-clip干扰)。 soft-clip干扰表明reads末端存在低质量核苷酸或,造成无法比对的干扰。 STAR会手动尝试剪掉未对齐的部分,只保留对齐的部分,不允许soft-clip storm。 在平均比较速度方面,分别比STAR和STAR快2.5倍和约100倍。
2. 解理位点检查与评价
转录组或RNA测序获得的reads与DNA测序的区别在于reads可能来源于两个(或多个)外显子区域,导致reads的一端在比对时与第一个外显子对齐。 前面部分,另一端与第二个外显子的后面部分(如右边的reads)进行比较,然后产生切点(site),带有site的reads称为reads,有a on the 剪接、鉴定、替代剪切分析和差异分析都非常重要。
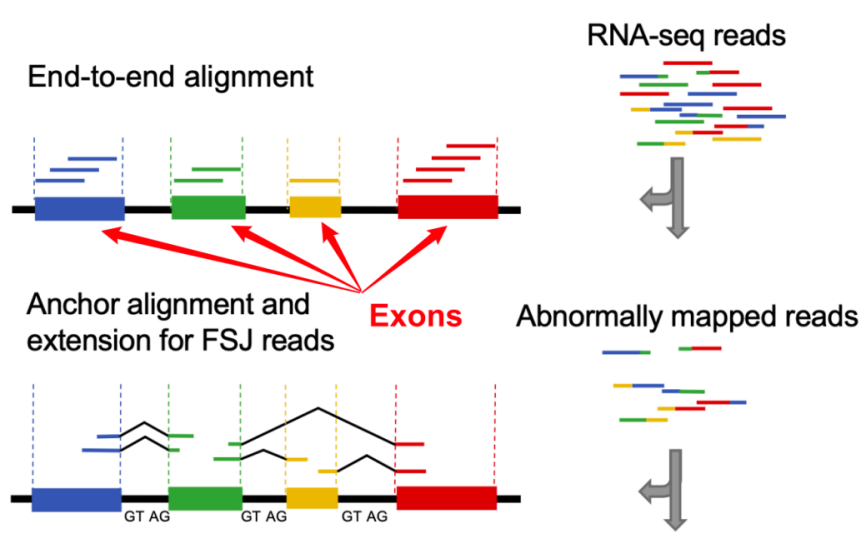
研究人员使用维恩图显示了不同比较工具测量的共同和独特的切点(如右图所示)。 数字代表刀具测得的切割点数,百分比代表每组切割点数。 验证比率。 dbEST 数据库中至少有两个表达序列标签支持的站点被用作阴性对照。
结果显示所有样品中拼接点验证率最高 (80%-91%),尽管测量或预测的拼接点总数明显多于 STAR。
3. 基于比对的转录组组装评价
在基于剪接的比对之后,转录组组装可用于识别表达的转录本组。 研究人员比较了两种最广泛使用的用于下一代测序数据的转录组组装工具和 . 对于比较部分, , STAR 和 。
除了short-read 检测方法,研究人员还对IDP( and )预测工具进行了研究。 IDP采用混合方法,利用短读长辅助长读长进行检测(与基于GMAP相比)。 将预测的亚型或转录本与 v19 中的参考转录组注释进行比较以测试准确性,并且 v19 中缺失的转录本被认为是假阳性 (FP),即假阴性。
通常,每个转录本中包含的外显子数量可以作为转录本组装质量的评价标准,单外显子转录本通常被认为具有最差的有效性。 从单外显子转录本的数量来看,约占30%,约占15%,此类单外显子转录本约90%为假阴性(FP)。 就拼接转录本的数量而言,比它多了50-200%。 IDP组装了所有的多外显子转录本(很难识别单外显子转录本),其外显子数量分布与v19更相似。 并且Iso-Seq算法有94%的单外显子转录本和77%的多外显子转录本缺失,反映出Iso-Seq方法在检查新转录本时具有更高的灵敏度,但False 较高。
对于基因水平的组装,IDP的准确性和灵敏度最好,IDP组装了更多的多转录本基因(右图b),比IDP更准确和灵敏。
对于转录本级组装,IDP 比其他工具准确 20%,灵敏度介于(更灵敏)和(稍不灵敏)之间。 基于短读长的组装工具中,转录水平的准确率平均比IDP高11%,转录水平的灵敏度高25%; 组装速度比IDP快60倍左右,比IDP快50倍左右。
4. 成绩单从头组装评估
当参考基因组或转录组缺失时,可以使用从头组装构建转录本。 研究人员评估了三种广泛使用的转录本从头组装工具 Oases 和 -Trans。
倾向于预测更长的亚型和更多的基因和转录本,Oases 仍然在所有样本中形成最高的 N10 到 N50 值,表明其在检查长亚型方面的优势。 -Trans 在高表达基因处有一个峰值,表明测量高表达亚型的强烈倾向,并且在比对质量(与参考的一致性比率)方面平均比 Oases 高 3%。 将构建的转录本与参考注释进行比较,-Trans和-Trans分别具有更高的内含子水平精度和灵敏度,Oases和-Trans在内含子链水平精度方面优于-Trans。
5.表达式的定量比较
传统的表达分析是直接将reads与参考基因组或转录组进行比对,然后估计转录本产量。 如果您需要检查新识别的转录本的产量,您可以使用转录组组装工具,例如和。 当只关注已注释基因的定量时,可以使用reads直接比对参考转录组,然后使用RSEM等工具提高生产力。 例如,经典的无参考转录组首先与基于从头组装工具的参考转录组组装在一起。
另一种基于转录本的量化方法是直接判断reads从哪些转录本开始,无需比对,在估算资源上更经济。 , , quasi- 和 是这种估计方法的代表,解决了每个reads是由哪个异构体产生的问题。
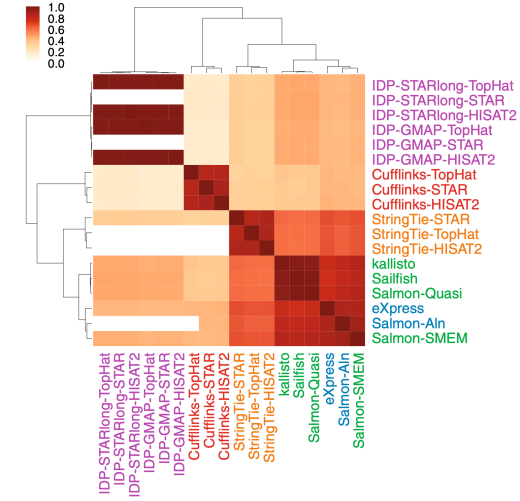
研究人员比较了基于基因组比对的定量工具(使用不同的比对工具)、基于转录组比对的定量工具与-Aln、无比对的定量工具、-SMEM 和-Quasi,以及基于长读长的 IDP(使用不同的短读长和长读长比较工具),利用上述组合得到一个样本的基因表达谱,对表达量取对数,进行秩和相关分析,评价表达谱的相似性。 结果表明,定量结果与其他工具的相关性最差(大于0.4),无需将直接定量工具与估计结果进行比较,相关系数为0.6-0.8。 -具有基于转录组比对的工具和 -Aln 的 SMEM 集群,并且 -SMEM 运行得更快。
研究人员还比较了同一样本的不同测序读数(MCF7-100 和 MCF7-300)的数据,以评估定量稳定性。 两个对齐无关的量化工具和-SMEM 具有最一致的量化结果。 总体而言,STAR-based比对结果的定量稳定性高于STAR-based比对,尽管作为短读比对工具,它在预测一致性方面最有效。 综上所述,non--based 是高效的,和 的组合是-based量化工具中性能最好的,但是速度比non--based工具慢了一个数量级。 通过比较不同的比较工具,研究人员认为在分析具有挑战性的样本时,Sum 优于 STAR。
6. 差异表达分析与评价
识别不同样本或不同处理条件下的差异表达基因集是许多RNA-seq的重要目标,检查差异表达基因的方法有很多,包括-based、limma和edgeR、-based和无需比较和量化用于差异分析。 在 SEQC 样本(SEQC-Avs.SEQC-B 和 SEQC-Cvs.SEQC-D)中通过 qRT-PCR 量化的 1001 个基因被用作对照来评估差异分析工具的性能。
在所有组合中,性能最好,而 、limma 和 edgeR 的性能稍差。 对于准确性, 和 的准确性仍然高于基于 的工具。 基于的工具比基于的工具效率更高,无需比对直接量化的工具可以获得高质量的差异分析结果。 对于 AUC-30 的恐惧,edgeR 表现最好,但与它相差不大。
7. RNA-seq 变异分析的评价
测量基因组和转录组变异对于理解基因表达的调控和可能影响基因表达的癌症相关变异至关重要,而 GATK 通常用于 RNA-seq 变异分析。 分析发现,与STAR不同的是,GATK和STAR在用于对比时具有相似的性能,总体上两者在不同样本上的执行时间相似。
8. RNA 融合检查评估
RNA-seq 的另一个重要应用是融合基因的测量,融合基因通常在各种疾病类型的发展中发挥关键作用。 常用JAFFA、STAR-、-、以及从RNA-seq中识别融合风暴。 除了基于短读长的分析工具外,IDP- 和 Iso-Seq 还可以从长读长 RNA-seq 数据中识别融合基因。
研究人员使用 MCF-7 卵巢癌细胞系中的 71 个经过验证的基因融合对该工具进行了评估。 具有最敏感和准确的预测,也表现出更高的灵敏度,而基于长读的 IDP- 表现出最好的准确性。
在运行速度方面,STAR-比其他工具快10倍以上,而and-估计资源需求更高。
总结:RNA-seq 分析中工具和估计方法的选择对分析的准确性和运行时间有很大影响。 具有最快和最准确的拼接对准,尽管不如 STAR 敏感,因此可能是涉及可变剪切剖面的管道的优先选择。 在大多数情况下,它比 . 更快、更准确。 和edgeR提供了最准确的差分分析dnastar拼接序列,可以作为差分分析的首选。 当用作比对工具时,GATK 同样适用于变异分析。
尽管缺少一些单外显子亚型,IDP 和 Iso-Seq 等长读长格式可以识别短读技术遗漏的许多新的多外显子转录本,这表明广度对于识别新的多外显子变异很重要。 然而,它在准确预测 RNA 融合干扰方面也具有显着优势,尽管可能会有更高的实验成本。 -SMEM 和其他无对齐工具具有最一致和准确的量化,前提是不需要检查新的异构体(可能只针对少数类型物种,它们具有相对可靠的异构体信息),-SMEM 可以用作最精确但资源高效的解决方案。
研究人员通过不同的工具组合对测试数据集进行了剖析,明确了哪种工具更适合转录组分析。 似乎从测试数据集的可以推导出在整体方面更有优势的各个工具的性能和工具组合,不一定完全适用于特定的数据集或目标基因我们很关心。 如果有兴趣并且有分析能力,对于个人来说,也可以考虑用不同的组合来分析同一个目标数据集,然后用实验验证的结果作为评价指标,对于大多数研究者来说是没有必要的。

参考:
SME,M,,H,PT,AuKF,,MB,,MP,E,.-RNA-seq.Nat.;8(1):59.doi:10.1038/-017-00050- 4.PMID:;PMCID:。
GaoY, WangJ, ZhaoF.CIRI:.Biol.;16(1):4.doi:10.1186/-014-0571-3.PMID:;PMCID:.
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码