
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-08-19
浏览次数:0
在上一篇文章中,我们过滤了数据质量控制,所以现在我的下一步是进行比较。 在进行比较之前,我们需要先了解一下比较的目的是什么? RNA-Seq数据比较和DNA-Seq数据比较有什么区别?
RNA-Seq 数据分析有多种类型,例如寻找差异表达基因或寻找新的替代剪接。 如果您正在寻找不同表达的基因,您只需要确定不同的读取技术即可。 我们可以使用bwa这样的比较工具,或者像这样的免对齐工具,但前者更快。
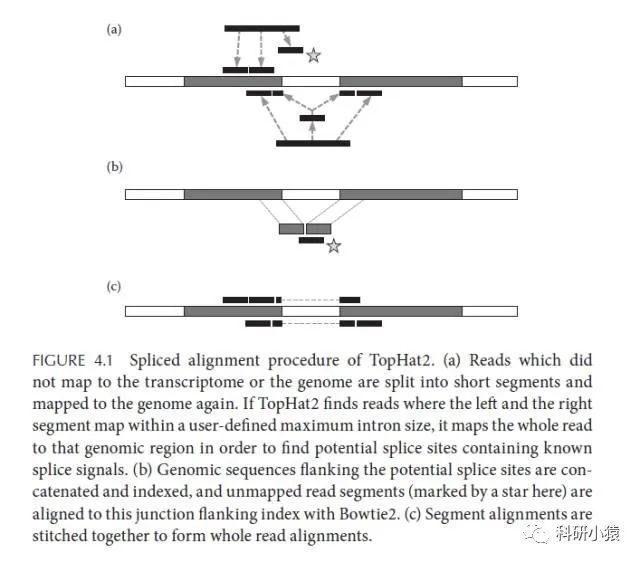
如果您需要寻找新的或替代的 RNA 剪接,您需要 STAR 等工具来查找剪接位点。 由于RNA-Seq与DNA-Seq不同,当DNA转录为mRNA时,内含子被部分去除。 因此dnastar序列比对,如果mRNA倒转的cDNA无法与参考序列进行比较,则会将其分离并重新比对,以确定中间是否有内含子。
本文的重点
下载索引
索引
比较
1.下载索引
通常有现成的索引可供人类使用。 我建议你尝试下载现成的,使用服务器自己创建索引,这会花费很长的时间。
#切换到工作目录,并创建index文件夹
master@master:~$ cd User/Projects/rna/biotree && mkdir index && cd index
#下载索引文件,并解压
master@master:~/User/Projects/rna/biotree/index$ wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/hg19.tar.gz
master@master:~/User/Projects/rna/biotree/index$ tar -zxvf hg19.tar.gz
2. 建立索引
工具上有一个命令是-build,只需要指定基因组fasta序列文件和创建的索引系列文件的前缀:
master@master:~$ conda activate rna
(rna) master@master:~/User/Projects/rna/biotree/index$ hisat2-build GRCh38.p13.genome.fa hisat2_index_GRCh38
请记住,创建的索引系列文件的前缀非常重要,后续比较实际上需要这个前缀。
3. 比较
# hisat2 -p 线程数 -x 索引 -1 转录组文件1.fastq -2 转录组文件2.fastq -S 输出文件.sam
(rna) master@master:~/User/Projects/rna/biotree$ hisat2 -p 10 -x /index/hisat2_index_GRCh38 -1 SRR11618610_1.fasta.gz -2 SRR11618610_2.fasta.gz -S output/SRR11618610.sam
#重复其他两个数据
在复现这种代码的过程中,你可能会遇到各种问题dnastar序列比对,也有可能是我的代码不正确。 这时候你需要有足够的耐心和思考,并且相信你会做到。

如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码