
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-11-12
浏览次数:0
在一篇文章中完成
转录组分析方法
- 5·30 -
总结一下比较合适
啦啦啦~今天是周末!
今天的推送来自粉丝投稿~一起来看看吧!
01
转录组()

定义:转录组的概念最早由etl.提出。 1997年[PMID:],指特定物种的特定组织或细胞中转录的所有RNA的集合。
应用:
①转录组可以揭示基因表达的时空动态,反映生物体特定细胞、组织或器官在特定生长发育阶段所有基因的转录表达水平;
②同时转录组常用于比较某一器官、组织或细胞在不同环境条件下的基因表达差异;
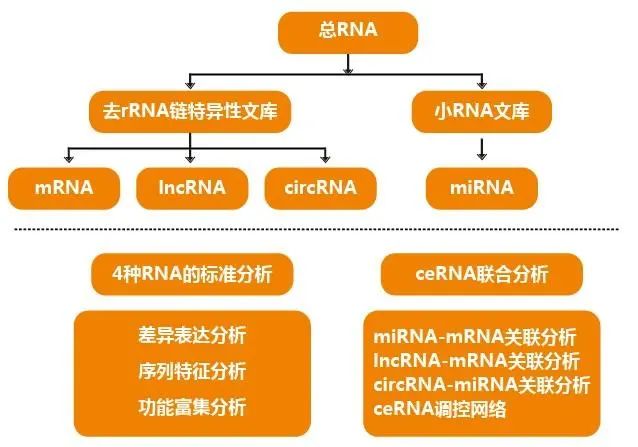
02
RNA-seq数据分析流程
RNA-seq,RNA,RNA 测序。
实验步骤如下:
① 靶组织总RNA的制备
② RNA样品的片段化
③ 反转录成cDNA片段
④ 构建测序文库
⑤ 原始数据处理(基于Linux或)
那么如何分析RNA-seq数据呢?
常规RNA-seq数据分析流程如下:
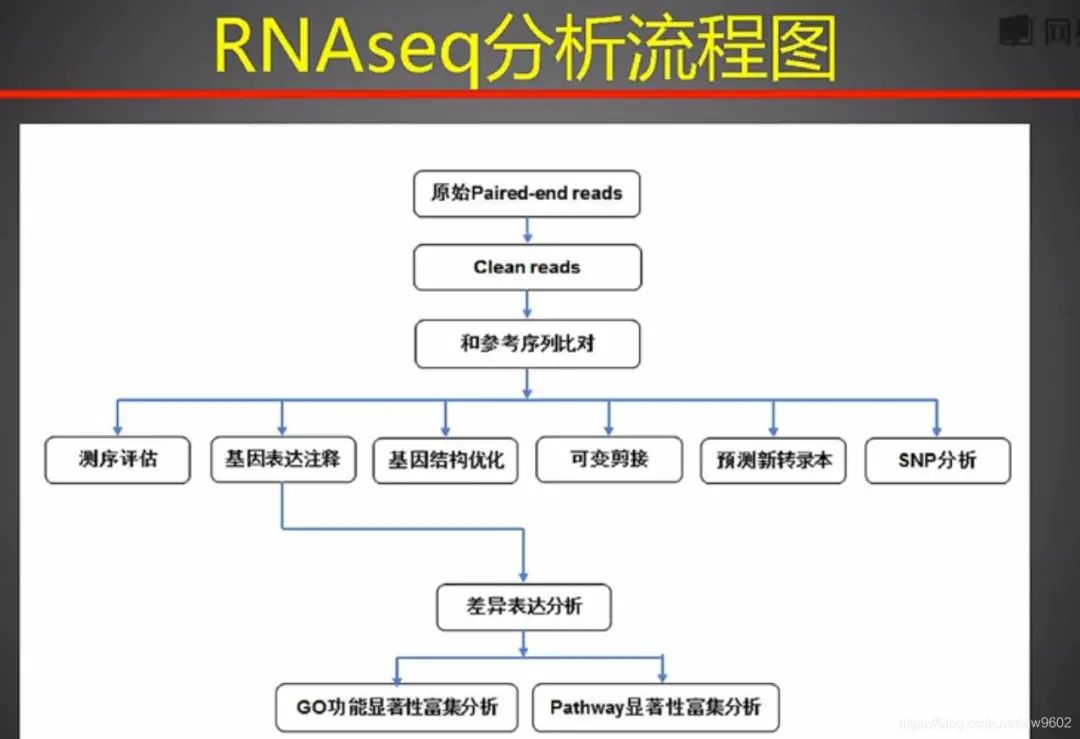
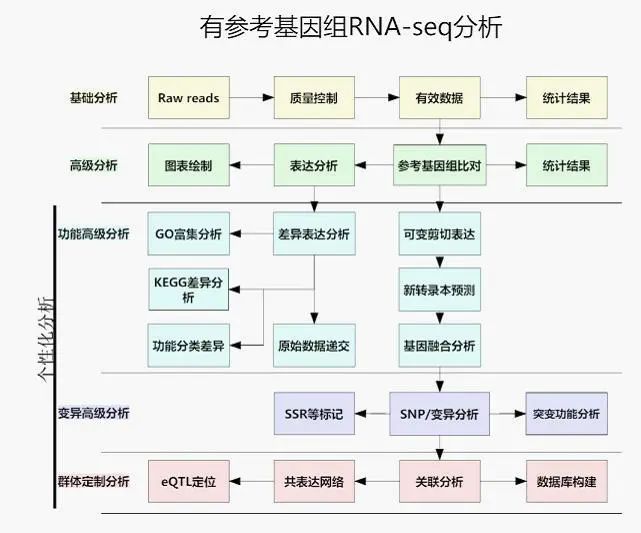
数字。 RNA-seq分析流程
下面,对分析动作的各步骤进行详细说明。
03
原始数据质量控制
① 原始数据格式:
序列确定后,以FASTQ格式输出原始数据;
② 序列质控软件:
序列质量控制软件主要包括:软件、软件等。
一)软件:
用于:过滤掉连接器()、低质量、重复和未检测到的原始数据序列,以获得干净的数据;
B)软件:
用于:检测干净数据的碱基质量值(score、Q-score)和碱基分布,确定测序和过滤效果,并用碱基测序错误率(Q20和Q30)来表示质量。
其中:Q20和Q30分别代表测序错误率为1%和0.1%的碱基比例。
GC 含量和样品之间的相关性也用于鉴定原始测序的质量。
04
Clean Reads 的对齐和拼接
完成从Raw Data到Clean Data的原始数据质量控制后,下一步就是Clean Reads的比对和拼接。
一般来说,对于不同类型的测序数据,组装序列和比较序列的方法有不同:
① 参考基因组序列测序数据:
当将测序数据与参考基因组序列组装时,您可以:
a) 首先通过序列作图()将所有reads定位到参考基因组;
b) 然后将映射到相应位置的reads进行聚类,形成代表所有可能的选择性剪接形式的图;
c) 最后将图信息转换为转录信息;
具体步骤如下:
A) 首先从UCSC数据库()下载参考基因组序列,并使用软件将其构建成基因组索引(index)文件。
B) 然后,在构建的索引文件、全基因组序列信息和参考基因注释信息的指导下,利用软件对基因组上的质控测序reads进行定位。
常用的序列比对软件如下:
,, STAR、GSNAP、SOAP2、S-MART 等
② 无参考基因组序列的测序数据:
在本领域,在没有参考基因组序列的情况下对测序数据进行序列组装也称为从头测序组装(de novo)。
基本流程是:
a) 将每个测序读段按照()的顺序拼接成重叠群;
b) 然后组装成括号();
c) 最后将脚手架中间的空隙填满,组装成一个长的连续序列;
d) 通过与模型动植物序列比对确定基因序列(基本本地工具,BLAST);
常用的序列比对软件如下:
以软件为代表的从头组装平台为转录组从头组装提供了有效的工具。
该软件包括 、 和 3 个独立模块。
使用时,三个软件模块按顺序运行,将读数组装成完整的转录本。 拼接策略是将干净的数据进行分割拼接,得到多个独立的demap(基因转录产物),然后通过reads溯源。 这些去图谱分类最终产生了全长转录本,并根据图谱分类解析了旁系同源基因。
从头开始组装的常用软件包括:
伊德纳
深渊
抄本的问题:
在真核细胞中,每个基因可以产生多个转录本。 这会导致同一个测序片段同时出现在多个转录本中,导致RNA剪接成多个不相连的图结构,每个图对应该基因相应的转录产物。
上述两种方法在进行序列拼接时,都希望将算法问题简化为各个基因的拼接问题,并提出了拼接图、重叠图、去图等多种图模型来实现各个图与基因之间的连接。相应的基因。 一对一对应。 两种方法采用不同的拼接策略,各有其使用范围,并且相辅相成。
一般来说,由于没有参考信息,且受测序误差、覆盖不均等因素影响,从头拼接算法的准确度明显低于基于参考基因组的拼接算法。 然而,有些物种并不具有完整的基因组序列,而从头剪接方法对于这些物种来说是开创性的。
此外,虽然有些物种拥有完整的基因组序列,但由于某些疾病等因素,其基因组发生了严重的突变和缺失。 在这种情况下,从头拼接方法将显示出明显的优势。
待解决问题:
可以说,尽管进行了几十年的研究,序列拼接仍然没有找到令人满意的解决方案。 主要问题如下:
A);
B) 序列组装的时间和空间挑战;
C) 序列片段错误(杂质序列和碱基读取错误);
D)之间的位置和距离无法准确确定;
上述问题往往会导致数据库出现不可避免的拼接错误。
05
转录本表达分析
构建的转录组分析方法可以对每个基因的表达进行归一化,定量估计表达,并根据映射到基因组的序列数量进行差异表达分析。
常用的软件有:
/,,RSEM,DESeq,edgeR
使用这些软件,您可以计算:
A) 表达基因映射的reads数量;
B) 基因长度和结构;
C) 获取基因的RPKM(reads per per reads)或FPKM(per of exon model per reads)值;
D)最后进行表达差异分析
一般来说,
A) 软件:可以从同一转录本中获取位于基因组上的片段;
B) 软件:这些片段可以组装成全长转录本;
C) - pare软件:将全长转录本与现有基因组注释文件进行比较,评估转录本构建,并根据已知数据库中的转录本信息定义构建的转录本的内含子、外显子和外显子。 基因间和其他区域;
d) 软件:每组样本都可以拼接得到各自的转录本信息,软件可以将它们合并成一个转录本集合,作为下一步差异表达信息分析的基础。
E)包含的软件:可用于计算两个或多个样品的基因表达量,并根据基因表达量对表达丰度进行统计分析和统计检验,以获得不同样品之间差异表达的RNA分子。
F) DESeq包:在这一步中,还可以使用DESeq包来进行不同处理之间的差异表达分析。 通过计算两个处理中表达量相等的同一基因的P值,然后利用多重假设检验对F值进行修正,修正后的P值可以用来检验差异表达基因的显着性。
06
选择性剪接分析
对于选择性剪接的分析,一般使用软件将reads与基因组序列进行比对,根据位置、长度和结构信息预测剪接体的类型,然后利用软件包对剪接体进行差异表达分析。
其中,可用的软件宝藏包括:
,,,, 等等。
目前转录组序列剪接位点识别算法主要有两种:
① 以基因组序列中潜在位点为中心的剪接识别:
常用算法:
优点:可以同时利用多机、多核CPU资源,并行运行,大大提高分析效率。
缺点:其剪接模式搜索依赖于基因组中潜在位点的预先识别,只能检测特定距离内的保守剪接模式。
②以reads分割和匹配为中心的剪接位点识别:
常用算法:
优点:完全以reads为中心,寻找剪接位点,不受保守剪接方式限制
缺点:由于基因组本身重复序列较多,测序过程中形成过多短片段,影响计算效率和匹配精度,降低剪接位点的可靠性。
常用的可变剪切数据库包括:
H-InvDB
MAASE
.php
欧盟-普莱斯
.com
07
基因功能富集分析
基因功能:是指代表某些功能特征和生物过程的众多基因功能的集合。
基因功能富集分析:数百或数千个具有不同功能的基因、蛋白质或其他分子可以聚集到不同的生物通路中。 其中,往往富集了生物体中实现某些功能性状的一组基因或蛋白质。 到某条路径。 基因功能富集分析降低了后续分析的复杂性,还可以发现在生物过程中起关键作用的生物通路,有助于揭示和理解生物过程的基本分子机制。
常用的基因功能数据库包括:
GO、KEGG、、、、、、ERGO、Gen-MAPP
例如:
① KEGG数据库:
概述:可以系统分析细胞代谢途径和基因产物的功能,通过KEGG分析实现表达基因的富集,并获得相应的注释。
方法:该分析可以通过KOBAS或DAVID等软件来实现。 KEGG 网站为每个生物途径提供了专门的插图。
② 数据库:
概述:本数据库提供生物通路绘图模板,研究人员可以根据数据库要求和规定标准上传相应的生物通路分析结果。
但数据库不负责对用户上传的生物通路分析结果进行质量检验,导致资源质量参差不齐,数据库重复较多。 尽管如此,该数据库拥有海量的数据资源,并且与KEGG数据库不同的是,它包含了大量代谢途径以外的生物途径dnastar序列比对,这对于筛选能力较强的研究人员具有重要的参考价值。
③ 数据库:
概述:该数据库可免费使用。 用户可以利用数据库资源绘制自己的生物通路,并以MAPP格式保存。 这个文件很小,很容易在互联网上传播,有利于研究人员之间随时交流。
08
蛋白质相互作用网络
蛋白质相互作用网络
(-,PPI)
概述:蛋白质相互作用网络可用于分析差异表达基因蛋白参与的生物信号传递、基因表达调控、能量和物质代谢、细胞周期调控等生命过程。
意义:不仅可以了解蛋白质之间的功能联系,还可以了解疾病等特殊生理条件下生物信号和能量物质代谢的反应机制。
常用的数据库包括:
SWISS-PROT,,PDB,SDSPB等
软件:
将差异表达基因数据导入软件可以对蛋白质相互作用网络进行可视化分析。
对于数据库中没有注释信息的物种,需要将候选基因序列与数据库中包含的密切相关物种的基因进行比较,然后利用现有的蛋白质相互作用关系来预测候选目标基因之间的PPI数据库中的物种。 。
该方法首先需要根据文献信息确定候选分子之间是否存在或可能存在相互关系,然后利用软件构建交互网络并分析关键节点,最后确定PPI。
网络系统生物学可以利用网络分析方法从整体层面研究生命体中各种相关生命活动之间的相互关系和动态变化,为全面揭示生命奥秘提供重要的研究方法。 通过网络系统生物学的综合分析从文献挖掘中获得的信息弥补了还原论分析方法的缺陷,更有利于更深入地理解生命现象的科学本质。
09
加权基因共表达网络分析
加权基因共表达网络分析
(基因协同,WGCNA)
概述:WGCNA可以识别共表达基因、基因网络与表型之间的相关性以及基因网络中的核心基因。
目的:适用于不同器官或组织的发育调控、同一组织不同时间的发育调控等复杂数据模式的转录组数据挖掘。
方法:
两个环节:WGCNA分为两个环节:表达聚类分析和表型相关性;
四步:WGCNA的实施包括四个步骤:基因间相关系数计算、基因模块确定、共表达网络构建、模块与性状关联;
通过上述网络分析,我们可以找到调控中心的枢纽基因,进而深入分析该基因,探究其功能。
传统方法的缺点:
由于传统的表达调控网络分析方法是基于基因间表达的相关系数,当样本数过少时,相关系数不可靠,得到的调控网络价值不大。
WGCNA的优势:
WGCNA放弃了通过设定相关系数阈值来判断基因是“相关”还是“不相关”的做法,而是对基因之间表达的相关系数进行适当的加权,以避免信息丢失,并使相应的基因表达调控网络近似无标度。 网络分布。
10
转录本数据分析中的两个问题
在上面的讨论中,我们简要回顾了分析转录组数据的工作流程以及基本分析中包含的各个组件。
事实上,在分析转录数据时有两个重要问题:
① 如何从海量测序数据中准确拼接RNA序列;
② 如何从海量RNA序列中筛选目标候选基因并发现新基因;
下面,我们重点介绍一些筛选目标候选基因和发现新基因的方法:
11
序列同源性比较
基本假设:
如果基因A与基因B同源,则基因A可能与基因B具有相似的功能。
基本方法:
对转录组数据分析得到的差异基因进行富集分析,找到相关通路中富集的候选基因,然后在NCBI上找到该基因或蛋白序列,然后利用序列同源性比较来预测新的基因功能。
利用同源比对算法在DNA或蛋白质序列数据库中寻找待测序列的同源基因,获得一系列与该基因同源性较高的基因或片段。 这些基因或片段的已知功能信息对于进一步的研究是有用的。 该基因功能提供了指导。
实施方式:
① 同源搜索:
软件1: 软件:
可以通过软件进行同源基因检索。 序列检索时,软件根据序列的结构和可能的功能,通过直系同源基因和旁系同源基因来搜索待检索的序列。
一般认为,位于同一功能域的基因序列,即使来源于不同物种,也可以定义为直系同源基因;而属于不同功能域的基因序列,即使来源于同一物种,也可以定义为旁系同源基因。 同源性搜索可以为基因组进化分析提供证据并预测候选基因功能。 但基于该软件的直系同源基因搜索必须在相对完整的基因组之间进行;
软件2:HMMER 3.0软件:
此外,HMMER 3.0软件还可用于基于隐马尔可夫模型检测序列比对结果中的保守区域,识别序列中已知的核酸或蛋白质结构域,并阐明序列之间的超科、科、亚科和种。 特异性和其他关系。
优点是可以检测远距离同源序列。
同源搜索为发现进化或物种分化过程中的关键功能基因提供了重要工具。
② 多序列比对:
它同时对多个序列进行同源性比较,发现它们共同的结构特征。 该方法为寻找基因家族或蛋白质家族的保守区域提供了重要的技术支持。 保守区域与家庭成员的功能密切相关。 通过这些方法建立的蛋白质家族数据库可以帮助寻找新基因所属的蛋白质家族及其保守区域,并提供该家族其他成员的结构和功能信息。
多序列比对软件主要包括:
/X,, ,SAGA,MEGA7,MSA,MAFFT 等
这些多序列比对软件是使用各种程序开发的。
其中,应用最广泛的比较程序是基于渐进比较算法的Clust-alW程序,但其准确度不高。
因此,陆续开发了其他比较方案,其中较有代表性和影响力的有:
基于迭代求精策略的PRRN/PRRP;
基于傅里叶变换;
基于多次迭代和 T-COF-FEEJ 等。
这些新程序提高了多序列比对的速度和准确性。
12
选择信号方式
选择信号():
物种形成经历了自然选择或自然与人工的双重选择。 在长期的定向选择压力下,生物体的某些性状和表型会发生定向变化,引起基因组某些区域的遗传组成发生质和量的变化,导致不同等位基因频率的变化,甚至产生新的等位基因。突变。
其中,选择在基因组中留下的印记称为选择信号( )。
使用选择信号法挖掘功能基因转录组数据的步骤如下:
①首先使用Gatk或Gatk等软件筛选出转录组测序得到的SNP()数据;
②然后结合XP-EHH(交叉)方法检测SNP数据,得到群体选择信号;
③然后通过AND方法扫描群体选择信号,最终获得相关性状的基因组选择区域。
其中,由于每个选定区域的基因较多,因此首先计算每个基因中每个SNP位点的XP-EHH得分,然后将XP-EHH得分从低到高排序,得到该SNP位点的得分确定得分最高的基因所受到的选择强度,最后确定该区域中XP-EHH得分最高的SNP位点的得分作为所选区域所受到的选择强度。 因此,该区域中XP-EHH得分最高的SNP位点所在的基因就是该选择区域的候选基因。
构建系统发育树,对筛选出的候选基因进行分析,发现与物种群体和目标性状密切相关的筛选基因。 同时与自然选择和人工选择联系起来,进一步解释所选基因在当时环境和进化中的作用。
13
极端数据法
极端数据法基于以下假设:
健康组织中高表达的基因对于生物体充分发挥其生理功能具有潜在的重要作用。
在实际应用中:
采用百分位数法根据同源基因集的表达水平来划分:
高表达组:表达量位于前10%的基因集;
中表达组:位于基因集的10%~50%;
低表达组:位于基因集的50%~90%;
极低表达组:基因集位于底部10%;
其中,极低表达组和高表达组被定义为极端数据集。
极端数据方法是指:
比较物种A高表达组和物种B相应组织极低表达组共有的同源基因,两者的交集就是物种A表达的同源豪华基因,即该基因高表达在物种 A 中,与物种 B 中的相对应。组织中的表达非常低或没有表达。
类似地,可以筛选出在物种B组织中特异性高表达但在物种A相应组织中表达极低或无表达的同源基因。
目前dnastar序列比对,百分位数法已广泛用于挖掘相对高表达或物种特异性表达的同源基因。 但该方法也由于测序平台、比对方法和软件的差异,容易出现一定的系统误差,导致假阳性结果比例较高,影响基因挖掘的准确性。 高表达基因分类标准不适当也会影响该方法的准确性。 开挖效果。
14
G0注释和KEGG富集
基于GO和KEGG数据库开发:
David、GOSim等软件可以实现差异表达基因的注释、富集分析和功能预测。
GO分析:
一般认为,一组具有相同生物学功能和调控机制的基因具有相似的G0注释项。 只要计算已知基因和候选基因之间GO项的相似度,就可以推断出候选基因产物的近似函数。
通过比较已知功能基因与GO语义的相似度,为未知基因功能的预测提供重要参考。
DAVID、GO都是GO富集分析常用的数据库。 他们可以通过GO对差异基因进行分类,并基于离散分布分析分类结果的显着性、误判率和富集度,得到与实验目的显着相关的结果。 对假阳性率低、富集度高的基因功能进行分类,然后从这个分类中找到属于同一GO功能分类的变化基因。 通过检验其统计显着性,我们可以得到改变基因的主要生物学功能。
KEGG分析:
它计算差异表达基因通路的超几何分布概率,富集每个差异表达基因通路上的所有差异表达基因,然后利用统计检验确定与某些差异表达基因显着相关的通路,进而找出差异。 可能涉及表达基因的细胞生化过程。
通路富集分析的生物学假设是:
通路中上游基因表达的变化会导致下游相关基因表达的变化,从而改变该通路中大量基因的表达,并且表达的变化达到了富集分析的统计显着水平。
在众多的差异表达基因中,许多基因在相应的通路中彼此之间并不存在直接的调控关系,而是共同参与某一过程的不同环节。 这些基因大致构成了通路的整体轮廓。
实际分析中:
将差异表达基因集导入通路分析软件,通过超几何分布概率计算和基因差异表达显着性检验,预测差异表达基因显着富集的通路。
当然,预测结果需要深入观察和了解某个核心通路中基因的相互作用,以确定差异表达的基因是否具有生物学意义。 一些无关紧要的通路也值得从功能注释的角度进行解读,只要其结果可以解释,具有生物学意义,也可以作为后续生物学验证分析的候选基因。
KEGG等数据库收集了现有的研究成果,但许多通路信息还远远不够完整。 因此,很多途径只能显示粗略的调控途径,尚不清楚涉及哪些转录因子以及是否产生其他代谢产物。 知道。
数据库中这些通路的完整性也会影响富集分析的结果。 随着研究的深入,每条通路中越来越多的节点将被揭开,生命奥秘的面纱将逐渐被揭开,后续的差异也将被揭开。 基因功能分析也将变得更快、更准确、更高效。
15
BSR-Seq方法
聚类分离分析 (BSA)
它是将两个具有极端表型差异的目标性状亲本进行杂交。 F1代自交后,得到性状分离的F2代。 选择来自 F2 代中具有极端表型差异的个体的组织样本并混合以构建两个 DNA 或 mRNA 库。 在两个库之间,基因序列差异片段是可能存在目的基因或数量性状基因座(QTL)的候选区域。
该方法主要适用于:
定位物种质量性状的单基因或数量性状的主基因是快速获得目标性状主基因或与其密切相关的分子标记的有效方法,但对于次效基因的定位和分析意义不大。
BSR-Seq方法:
它将BSA与RNA-Seq相结合,实现实验设计、测序分析、差异表达分析、基因功能分析和鉴定的全链条设计。
该方法首先选择隔离群体中具有极端性状的个体,收集组织样本构建两个差异表型样本池,分别提取总RNA,并进行转录组测序。 根据测序得到的碱基总数(bp)与物种基因组大小的比例决定测序倍数。 将转录组测序获得的干净读段与参考基因组进行比较,将与参考序列的独特位置比对的读段用于SNP发现。 然后利用经典的贝叶斯算法对SNP位点进行分析,最终找到与突变表型密切相关的基因组片段。
大量的RNA-Seq数据确保了特定物种在特定表达模式下性状差异表达相关基因的SNP标记的开发。 通过对相关基因表达的进一步精细作图和差异分析,可以逐步鉴定候选基因及其功能。 但该方法的定位结果是由物种亲本多态性、深度测量、混合池数量等多个参数决定的。 如果想要获得最佳参数,需要进行多次模拟实验以及参考基因组的支持。
完成~
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码