
友好定价、专业客服支持、正版软件一站式服务提供
工作日:9:00-22:00

发布时间:2023-08-02
浏览次数:0
作为一个对微生物基因组了解不多、接触微生物基因组时间较短的人来说,这样系统的基因组学自学是一个漫长且极其困难的过程。 写下这个过程更是痛苦。
但生活中却有很多令人沮丧的事情。 人无远虑,必有近忧。 毕竟到了我这个年纪,我的担心就更多了。
好吧,如果我说太多的话,就会影响我拔剑的速度。
基因组学、转录组学、宏基因组学,如果我们想了解分析步骤,就需要了解建库的原理。 这里我集中讲一下二代测序。
基因组学是测量单个基因组,宏基因组学是测量样本中所有物种的基因组,转录组是测量mRNA(建库时使用cDNA)。
根据我们的分析目的,我们会进行两种分析,一种是重测序,另一种是从头测序。
重测序,这些情况对我们来说指的是基因组,我们想知道手头的样本的变异情况,就是这样。
重测序,这些情况都是针对我们没有参考基因组,需要进行基因组组装,然后进行基因预测和功能分析。
不过,无论哪种情况,有些步骤是相同的,那就是登陆数据的质量控制。
并且(连接的程序,还有很多其他程序,我用这个来学习,这就是我学到的)
这是一长串图片,可以帮助我们检查质量。 通常车外数据都会有自己的质量控制,太差的数据不会给客户dnastar拼接序列,除非客户数据有问题!
基因组测序的解剖
一般来说,最好的方法是二代+三代测序。 三代测序的读长长,二代测序准确,两者可以相互校正。
然后我们一般都会见面,更多的是二代测序。 首先,我们描述有参考基因组的情况。
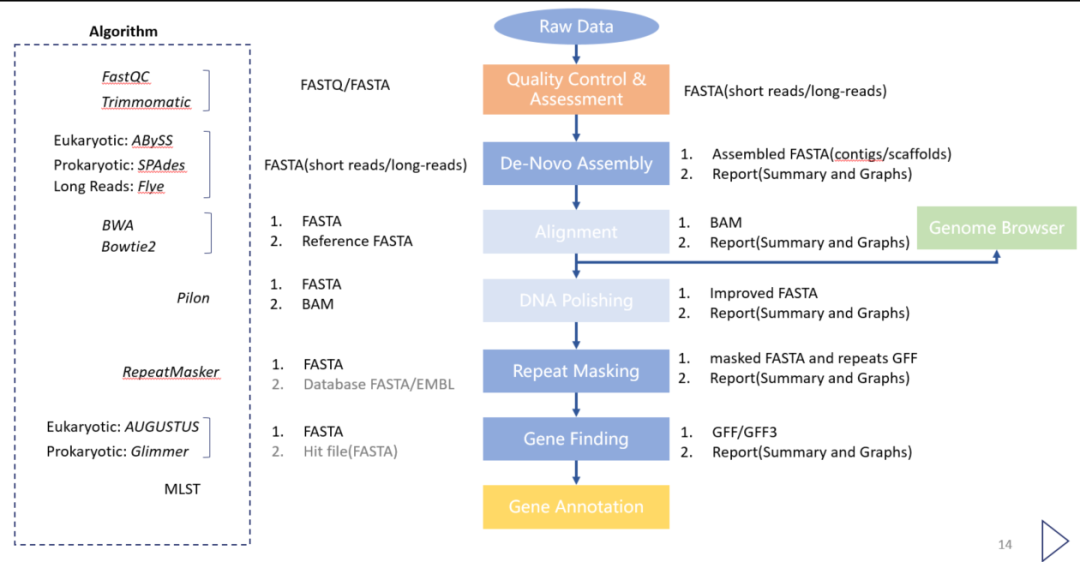
这样的基因组不需要基因组剪接,直接利用短读长来绘制参考基因组图谱。 常用的程序有BWA和2.输出BAM文件,可视化需要专门的可视化浏览器。
只有这个结果才能找到突变位点,SNP,indel,SV等,有些粗糙。 而这个结果还需要进一步解释,比如编码区实际上是否是非编码区,在此基础上还可以做进一步的解释,比如突变频率、致病性等,所以挖掘信息的空间是比较大的。
简单说一下两种比较程序的区别:BWA主要用于与参考基因组差异较小的短序列进行比较。 其中,BWA-MEM用于比对测序读数或将其组装成小型参考基因组,例如人类参考基因组。 该算法对测序错误具有良好的稳定性,适用的reads宽度范围广,从70bp到几Mb。
是一种超快速且节省内存的工具,用于将测序读数与长参考序列进行比对。 适合比对约 50 至 100 个碱基的读数。
现在我们来说说没有参考基因组的情况。
在没有参考基因组的情况下,如果我们想要挖掘信息,就需要重新组装基因组。 从头开始拼接的程序有很多,ABySS、Flye等等。
这是我的总结:
拼接策略
算法
优势
深渊
盛开
增加了整体视频内存要求,以允许组装小基因组。
图形
处理和数据、使用和读取的能力提供了混合组件。 它专为小基因组而设计,允许组装单细胞 MDA 数据以及标准分离株。
弗莱
图形,
用于单分子测序读取的从头组装程序,例如 和 。 适用于各种数据集,从大型真菌项目到小型饲养厂组件。
组装的序列可以直接用于预测基因和进行功能分析吗?
不,还有两个步骤,修正(针对三代:Pilon)和去重复序列()!
重复序列存在于多种物种中。 重复序列在真核基因组中更为丰富,例如,人类基因组的 47% 被认为由重复序列组成。 识别并掩盖重复且低复杂性的 DNA 序列,以改进下游基因预测。
别问我重复序列是什么,又是一个短篇故事,我们放张图吧!
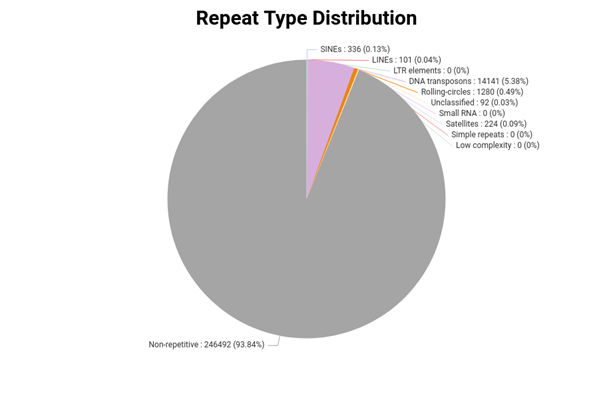
一般来说,对于重复的序列,不是直接删除,而是屏蔽掉,比如将序列改为大写。
屏蔽重复序列,好吧,出来进行基因预测。
这里分为真核基因预测和原核基因预测:
真核基因预测: .
是一种基于广义隐马尔可夫模型的真核生物基因预测软件工具。 为了预测基因组序列中不同区域和信号的统计特征,如内含子、编码外显子、UTR、启动子等。
包括100多个物种的预训练模型,如果分析的真核生物没有模型,则需要对模型进行训练,即需要将RNA-Seq、蛋白质、EST/cDNA等外部证据数据和数据进行训练。已上传。 提示是关于基因位置和结构的外在证据。 每个提示都是与特定基因组区域相关的本地信息。 在预测基因时,可以合并此类提示,这将改变候选基因结构的可能性。
它会倾向于预测与提示一致的基因结构。 输出结果包括三部分, 1. GFF 格式的预测基因组特征的坐标。 包含基因、转录本、内含子、起始密码子、终止密码子和CDS信息。 2.CDS序列:包含预测基因编码区核酸序列的序列表。 3.蛋白质序列:包含预测基因的蛋白质序列的序列表。
原核基因预测:
基于注册马尔可夫模型,它已成功用于寻找代表数百个物种的真菌、古细菌和病毒中的基因。 结果包括序列表,其中包含预测基因的核酸序列。 序列名称对应于行加上基因名称。
GFF3格式:在这里可以看到GFF文件的结果,包括序列ID、预测类型、起始和结束位置、正链和负链条件。
和常用工具一样,可以对剪接预测后的数据进行功能分析,比如蛋白质功能比对、GO注释、注释以及KEGG通路注释等。最后讲到功能分析的时候再一起讲。
对于基因组组装序列或者框架图或者仅仅是高质量的数据,我们可以在此基础上进行MLST分型。
MLST 是研究重要公共卫生支原体物种遗传多样性的有用工具,提供了便携式且可重复的分型系统。 它是一种基于核酸序列的方法,通常使用七个管家基因的内部片段序列来表征真菌分离株。 (需要选择参考物种、MLST 等位基因序列和从 .org 获得的概况数据)。
说了这么多基因组数据,我们来说说转录组数据的分析。
转录组数据分析
当我们进行转录组测序时,我们会测量此时细胞中的所有 mRNA 转录。
1. 对测序样本进行质量控制,过滤reads,剔除低质量核苷酸。 与基因组相同,跳过。
转录组有一个参考基因组(你可以选择几个参考基因组进行分析,对吗?)
与基因组参考类似,过滤后的读数直接与参考基因组进行比较(比较软件:STAR 和 BWA)。
STAR 用于比对小基因组,并有可能精确比对第三代测序技术中出现的长(数千个核苷酸)读取。 BWA适合简单的大规模转录组数据比较,例如真菌。 由于STAR支持剪接位点和融合reads检查,结果不仅统计匹配的reads、剪接位点的数量和类型,还统计错位、插入和gap统计。
比较结果是Bam文件。 统计与基因组比对的Reads分布,定位区域分为CDS(编码区)、(内含子)、(基因间区)和UTR(5'和3'非翻译区)。 在基因组注释相对完整的物种中,与CDS(编码区)对齐的reads浓度一般是最高的。 与(内含子)区域对齐的读数源自前mRNA残基或发生在选择性剪接过程中。由内含子引起的内含子保留干扰,以及与(基因间区域)对齐的读数可能是从新基因或新的非基因转录的。编码RNA。
如果没有参考基因组,则需要组装转录本 ()。
用于转录组组装的组装软件,基于DBG(De)组装原理组装出高质量的序列。
1):借助高质量序列建立K-mer宽度的短序列库,然后通过短序列之间的K-mer-1宽度延伸短序列,得到初步的拼接序列
2):通过序列降维,然后为每个类创建一个图
3):处理图,根据图中的Reads和配对的Reads找到路径,得到转录本
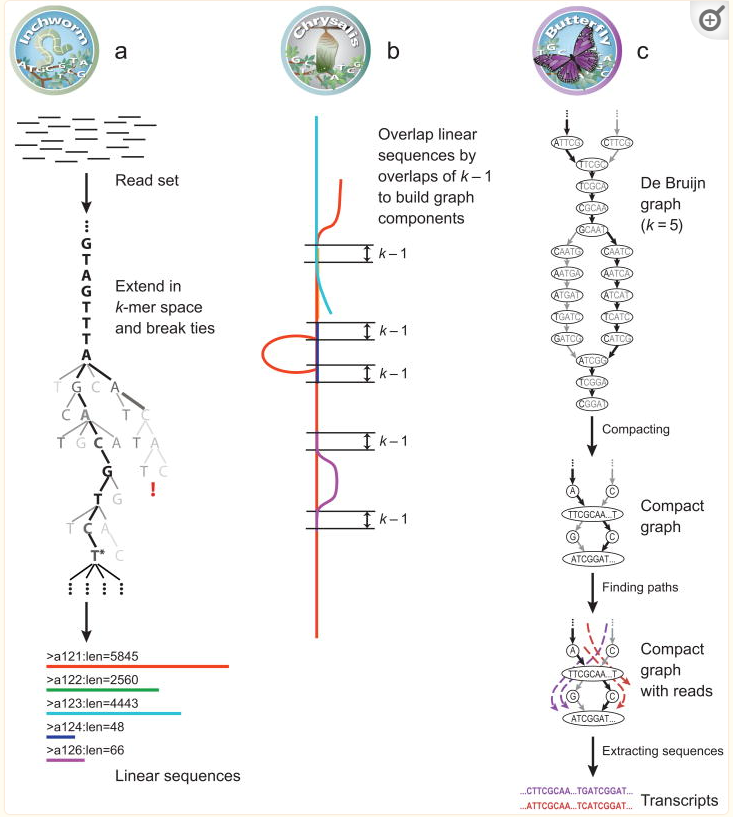
PMID 的:
虽然这个地方,拼接的结果有转录本/异构体。
我个人的理解:转录本是一个基因的多个转录本。
异构体,与该转录本对应的不同蛋白质(并非所有转录本都会被翻译。)
不过我看文献,据说是抄本拼接体……
剪接的下一步是进行完整性评估(BUSCO),顾名思义,即根据单拷贝基因来评估剪接结果的质量。
一般情况下,剪接后会进行基因预测,针对编码区(这里指的是ORF,ORF不是基因,也不是外显子)来预测转录组。
这里我也说一下CDS、ORF、外显子的区别,因为我老是犯错误:
CDS:CDS 是翻译产生蛋白质的 DNA 实际区域。 在原核生物中,ORF和CDS是相同的。
ORF:ORF 是一种 DNA 序列,以起始密码子“ATG”(并非总是)开始,以三个终止密码子(TAA、TAG、TGA)中的任何一个结束。 根据出发点,根据遗传密码将任何碱基序列翻译成多肽序列有六种可能的形式(正链上三种,互补链上三种),称为阅读框。 可能含有内含子。 CDS一定是ORF,而ORF不一定是CDS。
外显子:外显子是基因的任何部分,通过 RNA 剪接去除内含子后,将产生基因最终成熟 RNA 的该部分。 (摘自网络解释)
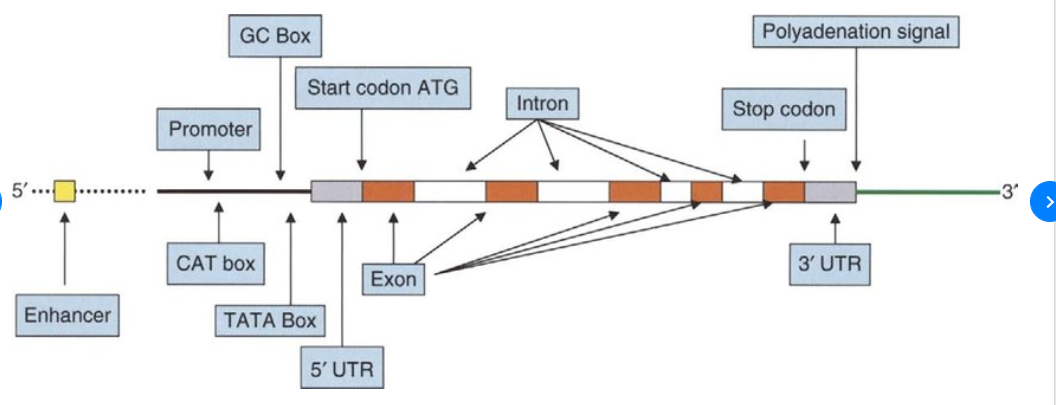
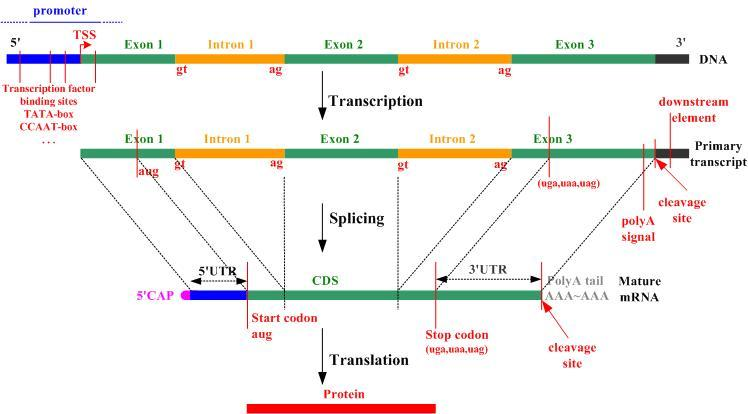
这是我在网上找到的两张图片,大家看一下。
我的理解是:ORF是多肽序列对应的mRNA或DNA序列,也就是说从起始密码子到终止密码子的mRNA序列(从mRNA上的AUG开始到终止密码子结束,或者DNA序列从 ATG 开始,以 TAA、TAG、TGA 结束)。 之后它可能包含内含子。 这是ORF,ORF可以翻译成蛋白质序列,所以这个ORF可能是一个基因,前一个基因的一部分,所以ORF越长越好。
成熟的 mRNA 剪接内含子,留下外显子(富含 5' 和 3' 非翻译区。)
因为CDS是只能翻译多肽的序列,所以不等于外显子。 就这样。
在转录组的编码区预测中,将输出GFF格式的预测CDS、预测蛋白和预测编码区坐标。
在进行下一步分析之前,会进行一个去冗余步骤,这里提到的是CD-HIT得到代表序列()。
接下来就是转录组分析最常见、最重要的目的——差异基因分析。
第一个是定量的。
对于参考基因组,可以使用 HTSeq 包。 采用HTSeq统计比较各基因的值,作为该基因的原始表达量。 读取计数与基因的真实表达水平以及基因的宽度和测序深度呈正相关。
这里将 BAM/SAM 文件与 GFF/GTF 注释参考文件进行比较。
下面是读取特征的方式,即这些比较的就认为是比较的,那些比较的就认为不是比较的,通常按照Union方案来统计。

在没有基因组的情况下,转录本序列只能作为参考,将每个样本的与参考序列进行比较,利用软件RSEM获得统计结果。
规划工作完成后,下一步就是进行不同的表达分析(针对给出的中学策略)。
得到的MA图、火山图、MDS图、热力图都是不同的表达可视化图,就不赘述了。
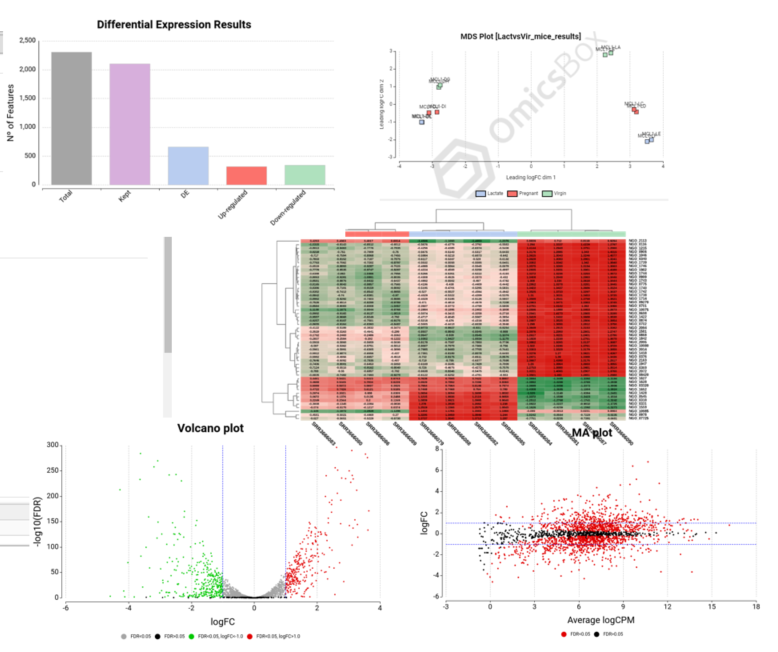
同样的,我们也可以对差异基因进行富集分析,上面也提到了。
这里顺便说一下三代转录组测序()。 由于三代reads较长,请勿拼接。
:每次测序运行均由 ccs 软件处理,为每个 ZMW(零模式波导)生成代表性的 CCS。
质粒清理和复用:使用 lima 执行质粒清理和条形码调用。
细化:此步骤包括 Poly(A) 尾部修剪以及多联体识别和去除。
降维:
(选修的):
剩下的就和二代分析差不多了,定量,分析……
宏基因组学
我想研究沉积物的人应该对此很熟悉。
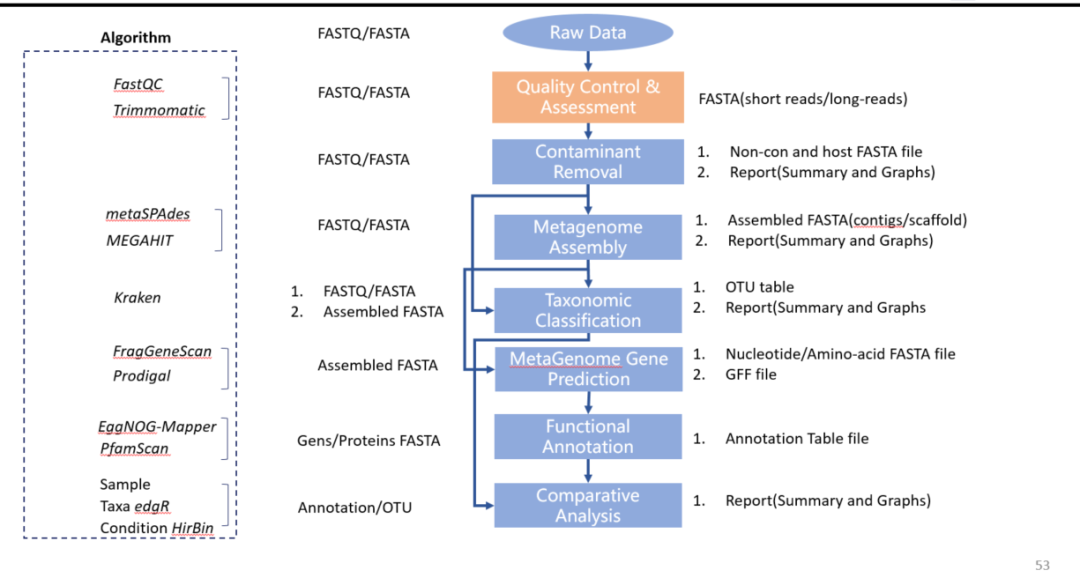
我们收到离面数据后,首先进行质检和拼接。 在获得高质量的序列后dnastar拼接序列,如果我们在进行宿主相关的研究,一般需要从测序数据中分离出宿主相关的DNA。
在自然环境中观察到的真菌中,只有不到 1% 可以在正常实验室条件下培养,这使得绝大多数真菌难以用传统微生物程序进行研究。 宏基因组学是将测序技术应用于自然环境中微生物群落的 DNA,从而测量此类样本中微生物及其基因的整体多样性。
宏基因组学实验的第一个主要目标通常是测量和量化存在的微生物。 这个过程称为分类分类或分析。 评估样本分类组成的主要策略有两种:扩增子测序 (16S/18S/ITS) 和全基因组测序 (WGS)。 扩增子测序分析虽然成本较低,但也有一些局限性,并且在某些真菌物种中,它们的 rRNA 基因之间没有足够的差异,无法进行物种鉴定。 宏基因组测序是来自整个样本群落的所有基因组信息,也可以识别物种。
分类
简而言之,通过物种组成进行 OTU 分类和产量分析。
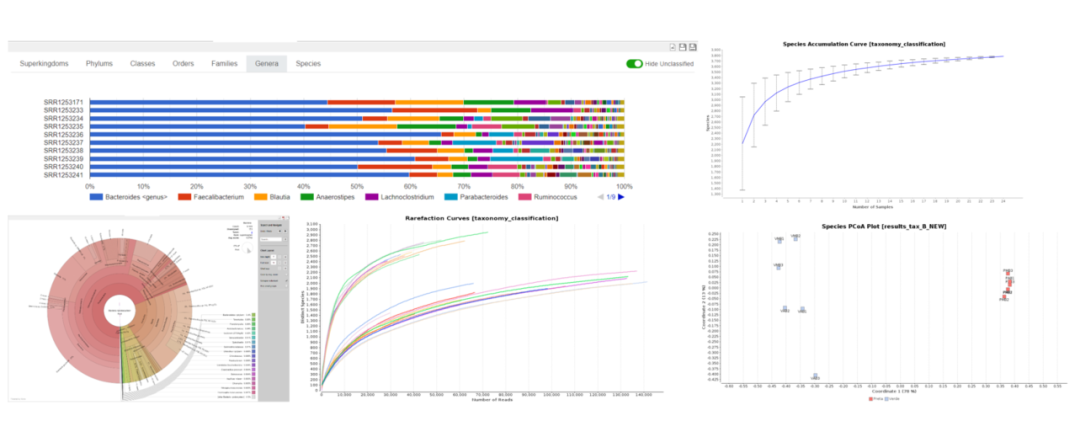
宏基因组组装:
拼接策略
算法
优势
德图
大型基因组设计需要更多的资源并花费更多的时间,但也会带来更好的结果,即更高的 Nx 值。
德图
用于以省时且经济高效的方式组装小型且复杂的宏基因组数据。
宏基因组基因预测:
是一个用于在短读段中查找(片段化)基因的应用程序。 它还可用于预测不完整组装或完整基因组中的原核基因。
是一款用于真菌和古细菌基因组蛋白质编码基因预测的软件。
结果输出:
接下来是功能分析:
-和
借助史密斯比对算法对旁系同源基因簇进行了功能注释。 旁系同源基因是指由于物种产生的进化过程而在不同个体中形成的同源基因。 这个基因起源于一个共同的祖先; 因此,在进化过程中,旁系同源基因一般都保留着相同或相似的特征。 - 是一种使用基于正交分配的预先估计快速对新序列(基因或蛋白质)进行功能注释的工具。
输入带有分析的序列(可以是蛋白质序列和基因组序列),蛋白质序列用于在整理数据库中搜索,猜测同源蛋白质,功能注释(参考链接:)。
输出结果还包括 GO 注释的链接。
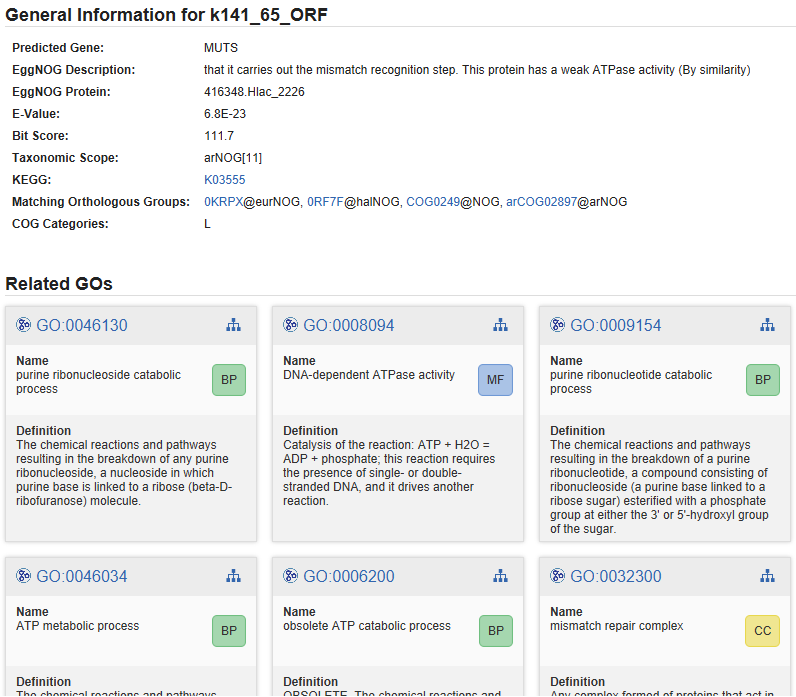
Pfam 是一种广泛使用的蛋白质家族域数据库,它依靠多重序列比对和隐马尔可夫模型 (HMM) 来识别一个或多个蛋白质功能域。 它是一个蛋白质比较工具。

宏基因组样本的比较与分析(分类产量比较和功能差异产量分析)
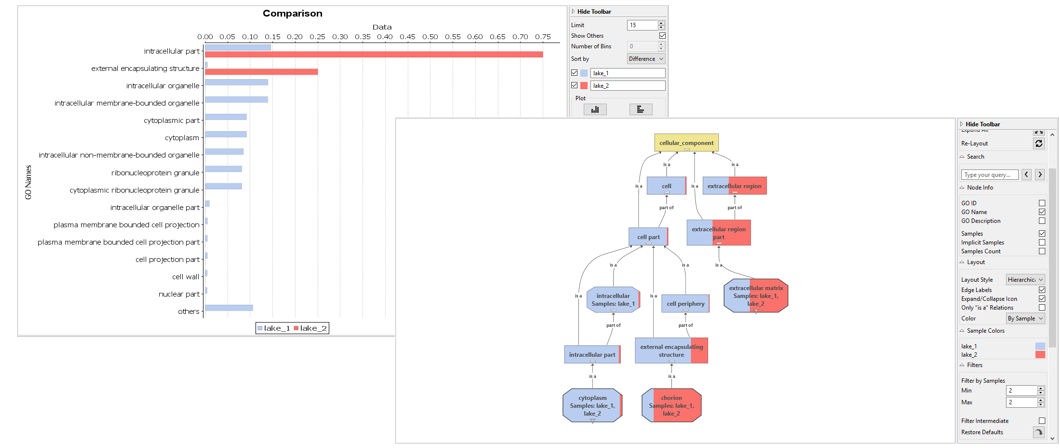
注释差异分析
差异产量分析
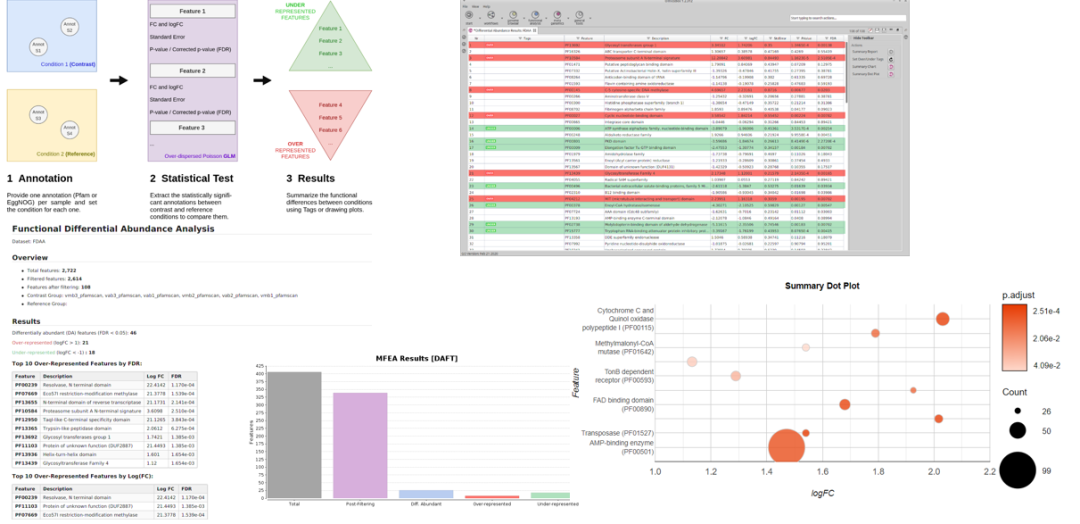
功能差异产量分析
接下来详细说一下功能分析,即用于功能注释和序列数据分析。 主要是技巧。 通过提取与获得的命中相关的 GO 术语并返回查询序列来评估 GO 注释。 酶代码是从等效的 GO 图谱中获得的,而基序是直接在 . GO注释可以通过重建基因本体关系和通路结构来可视化。 主要包括5个步骤:、、标注、统计分析和可视化。
如有侵权请联系删除!

官方公众号
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
Copyright © 2023 江苏优软数字科技有限公司 All Rights Reserved.正版sublime text、Codejock、IntelliJ IDEA、sketch、Mestrenova、DNAstar服务提供商
13262879759
微信二维码